
N° 1623
______
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
QUATORZIÈME LÉGISLATURE
Enregistré à la Présidence de l'Assemblée nationale le 11 décembre 2013.
RAPPORT
FAIT
AU NOM DE LA COMMISSION DES AFFAIRES SOCIALES SUR LE PROJET DE LOI portant diverses dispositions d’adaptation au droit de l’Union européenne dans le domaine de la santé,
(Procédure accélérée)
(Première lecture)
PAR M. Olivier VÉRAN,
Député.
——
Voir les numéros :
Assemblée nationale : 1336.
SOMMAIRE
___
Pages
INTRODUCTION 5
I. GARANTIR LE DROIT AUX SOINS TRANSFRONTALIERS 9
A. LE DROIT DES PATIENTS AUX SOINS TRANSFRONTALIERS 9
1. La reconnaissance progressive des soins transfrontaliers 9
2. Les apports de la directive du 9 mars 2011 10
B. L’AMÉLIORATION DE L’ACCÈS AUX SOINS TRANSFRONTALIERS 11
II. LUTTER CONTRE LE TRAFIC DE MÉDICAMENTS FALSIFIÉS 15
A. UN PHÉNOMÈNE MULTIFORME EN PLEIN ESSOR 15
1. La diversité des médicaments falsifiés 15
2. Le développement du trafic de médicaments falsifiés 16
B. L’INTERVENTION DES POUVOIRS PUBLICS 17
C. L’INCIDENCE DE LA VENTE EN LIGNE 19
III. CONFORTER LE SYSTÈME DE PHARMACOVIGILANCE 23
A. DEUX SYSTÈMES COMPLÉMENTAIRES 23
B. LA CONSOLIDATION DE LA PHARMACOVIGILANCE 24
C. VERS UNE ÉVALUATION CONTINUE DES MÉDICAMENTS 25
IV. UNIFIER LES RÈGLES EN MATIÈRE DE PRODUITS COSMÉTIQUES 27
Article 1er Responsabilité civile professionnelle des chiropracteurs et des ostéopathes 51
Article 2 Sanction du manquement à l’obligation d’assurance de responsabilité civile professionnelle des chiropracteurs et des ostéopathes 55
Article 3 (art. L. 5122-14, L. 5131-1 à L. 5131-11, L. 513-10-2, L. 513-10-3, L. 513-10-4, L. 513-10-5 à L. 513-10-10 [nouveaux], L. 5431-2, L. 5431-5, L. 5431-6, L. 5431-7, L. 5431-8 et L. 5431-9 [nouveaux], L 5437-2, L 5437-3 à L 5437-5 [nouveaux] du code de la santé publique) : Produits cosmétiques et de tatouage 56
Article 4 (art. L. 4 362-9-1 [nouveau] et L. 4363-4 du code de la santé publique) : Vente sur internet de lentilles correctrices 70
Article 5 (ordonnance n° 2012-1427 du 19 décembre 2012, art. L. 5124-1, L. 5125-33, L. 5125-34, L. 5125-39, L. 5438-2, L. 5438-6, L. 5438-7, L. 5438-8 [nouveau] du code de la santé publique) : Lutte contre la falsification des médicaments et encadrement de la vente en ligne par des pharmaciens d’officine 75
Article 6 (art. L. 5121-9-4 et L. 5124-6 du Code de la santé publique) : Information sur les motifs des décisions des exploitants de médicaments qui en suspendent ou arrêtent la commercialisation 85
Article 7 (art. L. 5121-1-2 et L. 5121-1-4 [nouveau] du code de la santé publique) : Harmonisation du contenu des prescriptions transfrontières 88
TABLEAU COMPARATIF 97
ANNEXE AU TABLEAU COMPARATIF 131
ANNEXE : LISTE DES PERSONNES AUDITIONNÉES 141
Le projet de loi portant diverses dispositions d’adaptation au droit de l’Union européenne dans le domaine de la santé vise à remplir l’obligation constitutionnelle de pleine application en France du droit européen.
Conformément à la décision du Conseil constitutionnel du 10 juin 2004 sur la loi pour la confiance dans l’économie numérique, cette obligation découle de l’article 88-1 de la Constitution qui dispose que « la République participe à l’Union européenne constituée d’États qui ont choisi librement d’exercer en commun certaines de leurs compétences en vertu du traité sur l’Union européenne et du traité sur le fonctionnement de l’Union européenne, tels qu’ils résultent du traité signé à Lisbonne le 13 décembre 2007 ».
L’adaptation au droit européen implique le plus souvent une retranscription fidèle et précise de dispositions que le législateur national ne peut pas modifier.
Il peut s’agir de transposer des directives, qui, selon l’article 288, alinéa 3, du traité de fonctionnement de l’Union Européenne (TFUE), « lient tout État membre destinataire quant au résultat à atteindre, tout en laissant aux instances nationales la compétence quant à la forme et aux moyens ». Leur transposition consiste dès lors pour l’État membre à adopter toutes les mesures nécessaires à son incorporation effective dans l’ordre juridique national, ce qui emporte l’amendement ou l’abrogation des dispositions nationales incompatibles.
Lorsque le droit européen est établi par la voie des règlements, ceux-ci, conformément à l’article 288 alinéa 2 du TFUE, ont « une portée générale. Ils sont obligatoires dans tous leurs éléments et sont directement applicables dans tout État membre ». Les règlements sont valables uniformément et intégralement dans tous les États membres, ce qui interdit à ces derniers de les appliquer de manière incomplète ou de procéder à une sélection parmi leurs dispositions
Le présent projet de loi a donc pour objet de traduire dans le droit national les objectifs fixés par plusieurs directives européennes : ceci n’est nécessaire que dans la mesure où les dispositions nationales n’y satisfont pas déjà. De manière plus contraignante, il vise également à adapter le droit national à un règlement européen : si les dispositions du règlement sont directement applicables, ses dispositions peuvent être reprises dans des textes nationaux à des fins d’accès au droit ; elles peuvent également être complétées, dans la mesure où le règlement l’autorise.
Les textes européens confèrent donc des marges de liberté : le projet de loi soumis au Parlement s’est efforcé de les utiliser.
Dans tous les cas de figure, une adaptation rapide et complète au droit européen est nécessaire. Lorsque la Commission européenne, gardienne des traités, constate que le droit national contrevient au droit européen, ou qu’il existe un retard dénué de toute justification dans la transposition des directives, elle peut initier une procédure d’infraction invitant l’État membre à mettre sa législation en conformité qui peut aboutir à un recours en manquement engagé devant la Cour de justice de l’Union européenne.
Dans ce cas, le traité de Lisbonne prévoit désormais que les États membres peuvent être condamnés à payer des amendes et des astreintes dès le recours en manquement et non plus seulement en cas de retard ultérieur dans l’exécution de la décision de la Cour, conformément à l’article 260, paragraphe 3 du TFUE. Dans une communication du 11 novembre 2010, la Commission indique qu’elle compte faire usage de cette nouvelle faculté « par principe dans toutes les affaires concernant les manquements ». S’agissant de la France, la Commission a fixé, en 2010, les montants minimaux de l’amende forfaitaire et de l’astreinte journalière à, respectivement, 10 millions d’euros et 12 134 euros.
En outre, tout retard d’adaptation du droit national porte atteinte au principe de sécurité juridique des citoyens européens : le maintien dans la loi de dispositions contraires au droit européen conduit ces dernières à être écartées par le juge national. Il appartient en effet aux juridictions administratives et judiciaires d’exercer le contrôle de compatibilité de la loi au regard des engagements européens de la France et, le cas échéant, de saisir la Cour de justice de l’Union européenne à titre préjudiciel.
Le juge peut ainsi être conduit à sanctionner un défaut de transposition de deux manières : par une annulation du texte de transposition en tant qu’il est infidèle ou incomplet1 ou par l’annulation d’actes administratifs conformes à la loi mais ne répondant pas aux objectifs fixés par des dispositions non transposées de directives ou en contradiction avec des dispositions précises et inconditionnelles qu’elles contiennent2.
Aussi peut-il être nécessaire d’intervenir, le cas échéant, en urgence, pour mettre la législation nationale en conformité avec le droit européen. Ce n’est le cas, dans ce projet de loi, que pour l’article 4 qui vise à mettre un terme à une procédure d’infraction engagée par la Commission européenne à l’encontre de la France en matière de vente en ligne de lentilles oculaires correctrices, le 27 juin 2007. Votre rapporteur renvoie, à ce titre, à ses observations au commentaire de l’article 4, ci-après.
Des dispositions ayant la même portée que celle de l’article 4 ont été adoptées, le 16 décembre 2013, par amendement, lors de l’examen en séance à l’Assemblée nationale, en nouvelle lecture, du projet de loi sur la consommation (article 17 quater). Les dispositions du projet de loi sur la consommation devant être promulguées avant celles du présent projet de loi, votre rapporteur estime préférable de privilégier ce vecteur législatif : ceci permettra d’écarter au plus vite tout risque de condamnation de la France. L’adoption de l’article 4 est donc inutile.
Les articles 1, 2 et 7 du projet de loi transposent les dispositions de la directive n° 2011/24/CE du Parlement européen et du Conseil du 9 mars 2011 sur l’application des droits des patients en matière de soins de santé transfrontaliers. Elles complètent le dispositif visant à garantir le droit de l’ensemble des citoyens européens de bénéficier de soins dans un État membre différent de leur État d’affiliation : ce droit est largement effectif en France, et les dispositions des articles 1, 2 et 7 approfondiront un mouvement d’intégration ancien auquel la France contribue au premier chef.
L’article 3 adapte les dispositions du code de la santé publique au règlement n° 1223/2009/CE du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques. L’unification des règles applicables aux produits cosmétiques améliorera le contrôle sanitaire tout en contribuant au développement économique d’un secteur particulièrement important en France.
En matière de médicaments, les articles 5 et 6 achèvent la transposition des objectifs fixés par deux directives qui ont modifié la directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant le code communautaire relatif aux médicaments à usage humain : respectivement, en ce qui concerne la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés, la directive 2011/62/UE du 16 mai 2011, et en ce qui concerne la pharmacovigilance, la directive 2012/26/UE du 25 octobre 2012. Ces mesures renforceront la lutte contre le trafic de médicaments falsifiés, encadreront strictement la vente en ligne de médicaments non soumis à prescription médicale obligatoire et conforteront les systèmes national et européen de pharmacovigilance.
Aussi, l’adoption du présent projet de loi permettra-t-elle de renforcer la contribution de la France au système de soins européen.
Au sein de l’Union européenne, les soins de santé transfrontaliers recouvrent l’ensemble des soins dispensés ou prescrits à un assuré dans un État membre différent de son État d’affiliation à la sécurité sociale. L’accès à ces soins est facilité par la directive n° 2011/24/UE du 9 mars 2011 dont le présent projet de loi achève de transposer les dispositions.
La libre circulation des personnes au sein de l’Union européenne est un des principes fondamentaux consacrés par les traités communautaires. Afin d’assurer cette liberté, il est apparu nécessaire de coordonner les régimes de sécurité sociale des différents États membres.
Le règlement n°1408/71 du Conseil du 14 juin 1971 et le règlement n° 574/72 du Conseil du 21 mars 1972 ont tout d’abord permis aux travailleurs ressortissants d’un autre État membre d’être affiliés au régime d’assurance maladie de leur État de résidence en disposant des mêmes droits que les ressortissants de cet État. S’appuyant sur le principe de libre prestations de services, la Cour de justice, dans ses arrêts Kohll et Decker du 28 avril 1998, a ensuite reconnu aux ressortissants communautaires le droit d’accéder aux soins dans un autre État membre et à être remboursés sans autorisation préalable émanant de leur État d’affiliation.
Les règles de coordination des systèmes de sécurité sociale des États membres de l’Union ont été profondément remaniées par le règlement n°883/2004 du 29 avril 2004 qui pose le principe de l’unicité de la législation applicable à un assuré. L’État d’affiliation est en règle générale celui de l’exercice de l’activité professionnelle. Toutefois, des règles spécifiques trouvent à s’appliquer pour certaines catégories de personnes. Les fonctionnaires par exemple demeurent affiliés à la sécurité sociale de leur État d’origine. Un dispositif particulier est également prévu pour les travailleurs détachés.
Les règles relatives à la prise en charge, par l’assurance maladie, des soins reçus dans un État membre autre que celui d’affiliation, ont été modernisées par ce règlement ainsi que par le règlement d’application n°987/2009 du 16 septembre 2009. Les règlements opèrent une distinction entre les soins fortuits, reçus au cours d’un séjour temporaire effectué dans un autre État membre pour des raisons non médicales, et les soins programmés, reçus dans le cadre d’un séjour temporaire motivé par un traitement médical.
Dans le premier cas, les soins sont pris en charge grâce à la carte européenne d’assurance maladie, qui remplace depuis le 1er juin 20043 les formulaires précédemment utilisés pour assurer le remboursement des traitements permettant à l’assuré de terminer son séjour dans des conditions médicales sûres.
Dans le second cas la prise en charge est assurée via des formulaires à retirer auprès de l’organisme d’assurance maladie. L’autorisation préalable de l’État d’affiliation est dès lors nécessaire pour obtenir le remboursement de la plupart des soins programmés. En vertu de ces règlements, les États membres étaient largement libres d’accorder ou de refuser cette autorisation, mais l’accord est automatique lorsque le traitement requis, quoique pris en charge sur leur territoire, ne peut y être dispensé à l’assuré dans un délai acceptable sur le plan médical.
La directive n° 2011/24/UE du Parlement européen et du Conseil du 9 mars 2011 relative à l’application des droits des patients en matière de soins de santé transfrontaliers prévoit l’instauration d’un cadre général pour faciliter l’exercice du droit aux soins transfrontaliers. Elle ne concerne pas les services de soins de longue durée, les transplantations d’organes, ni les programmes de vaccination publique.
De nouvelles exigences s’imposent ainsi aux États membres. En premier lieu, il revient à chaque État membre de désigner un ou plusieurs points de contacts nationaux pour les soins de santé transfrontaliers. Ces points de contact sont en relation avec les associations de patients, les prestataires de soins de santé et les assureurs de soins de santé. Ils sont chargés de fournir aux patients des informations sur leurs droits lorsqu’ils décident de bénéficier de soins de santé transfrontaliers, ainsi que les coordonnées d’autres points de contact dans les autres États membres.
L’État de traitement organise et fournit les soins de santé. Il veille au respect des normes de qualité et de sécurité lors de la prestation de soins, notamment par la mise en place de mécanismes de contrôle. Il assure également le respect de la protection des données personnelles et de l'égalité de traitement des patients non ressortissants de son territoire.
L’État d’affiliation prend en charge le remboursement de la personne assurée, à condition que le traitement reçu soit prévu parmi les soins remboursables dans sa législation nationale. Le montant des remboursements est équivalent à ce qui aurait été remboursé par le système de sécurité sociale obligatoire si les soins avaient été fournis sur son territoire, mais ne saurait excéder le coût réel des soins de santé reçus.
L’État membre d’affiliation a la possibilité de rembourser des frais connexes, notamment les frais d’hébergement ou de déplacement. Une personne assurée peut également bénéficier de remboursements dans le cadre de prestations de services à l’aide de la télémédecine.
En matière de prise en charge des soins programmés, l’autorisation préalable devient l’exception. De fait, la directive ne permet aux États membres de prévoir un système d’autorisation préalable que dans trois cas :
- pour une prestation nécessitant une hospitalisation d’une nuit au moins;
- pour des soins hautement spécialisés et très coûteux;
- dans des cas graves et spécifiques liés à la qualité et à la sûreté des soins prodigués.
Cette autorisation reste obligatoirement accordée lorsque le patient a droit à des soins de santé qui ne peuvent être dispensés sur son territoire dans un délai médicalement acceptable.
Afin d’accroître la sécurité des soins dispensés dans un autre État membre, la directive prévoit la possibilité pour les patients de déposer plainte et de demander réparation. Toutes les prestations de santé doivent par ailleurs être couvertes par une assurance en responsabilité ou une garantie similaire.
Enfin, la reconnaissance mutuelle des prescriptions médicales établies dans un autre État membre est consacrée.
Lorsque des assurés ou des ayant-droits français séjournent dans un autre État membre, ils peuvent accéder aux soins de santé ambulatoires ou hospitaliers qui s’avèrent nécessaires d’un point de médical, qu’ils soient dispensés dans le secteur public ou privé. La carte européenne d’assurance maladie permet d’accéder à ces soins dans les mêmes conditions que les assurés locaux : si la prestation est gratuite pour ces derniers elle le sera également pour les assurés français.
Si la prestation en nature est payante, l’assuré français doit faire l’avance des frais puis demander le remboursement :
- soit sur place auprès de l’institution compétente de l’État membre de séjour, selon les tarifs et les formalités en vigueur dans cet État ;
- soit en France sur présentation des factures et des justificatifs de soins, auprès de son organisme d’assurance maladie.
Lorsque les assurés ou ayant-droits français souhaitent se rendre dans un autre État membre afin d’y recevoir des soins, ils doivent le plus souvent obtenir de leur organisme d’assurance maladie une autorisation de prise en charge des soins. Si leur demande est acceptée, ils se voient délivrer une attestation « Droit aux soins programmés » qu’ils doivent présenter au prestataire de soins. Après avoir avancé les frais, ils peuvent adresser une demande de prise en charge soit à l’institution compétente de l’État de séjour, selon les tarifs applicables sur ce territoire, soit à son organisme d’assurance maladie. Ce dernier prend en charge les dépenses de santé selon les tarifs applicables en France si l’assuré en fait la demande.
Un système de créances et de dettes entre États membres permet de s’assurer que les dépenses de santé (programmées ou fortuites) d’un assuré soient in fine supportées par son État d’affiliation, quel que soit l’organisme à qui l’assuré s’est adressé en premier lieu. En France, le Centre des Liaisons Européennes et Internationales de Sécurité Sociale (CLEISS) rembourse aujourd’hui les prestations servies par les institutions étrangères à des assurés du régime français et recouvre les sommes dues par ces institutions pour les soins reçus en France par les assurés relevant de leur régime4.
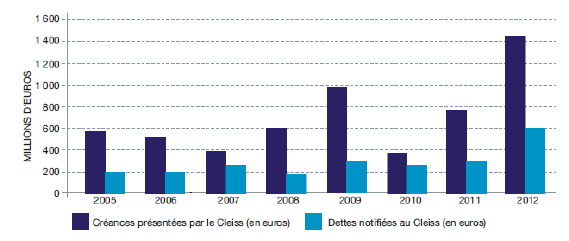
Entre 2011 et 2012, le montant des dépenses de santé remboursées par le CLEISS et les caisses françaises de sécurité sociale au titre des soins reçus par des assurés français dans des États membres de l’UE ou de l’AELE a augmenté de près de 75% pour atteindre 481 millions d’euros. Le nombre d’assurés français concernés par ce dispositif a lui aussi connu une hausse importante (+ 90%) entre ces deux années.
Les seuls remboursements effectués par le CLEISS ont été multipliés par 2,3 entre 2003 et 2012 avec de fortes irrégularités selon les années, soit un taux de croissance annuel moyen de 8,8% sur la période. Plus de 82% des remboursements effectués à des organismes étrangers en matière de soins de santé ont été reçus dans l’Espace économique européen ou en Suisse.
En 2012, les différents pays membres de l’Union ont versé 615 millions d’euros au CLEISS au titre du remboursement des soins reçus en France par leurs assurés, soit une augmentation de 10,6% par rapport à 2011. Entre 2003 et 2012, le montant ainsi remboursé au CLEISS a été multiplié par plus de 1,5, soit un taux de croissance annuel moyen de 4,5 %.
EVOLUTION DES CRÉANCES ET DES DETTES PRÉSENTÉES ENTRE 2005 ET 2012
Source : rapport statistique 2012 du CLEISS
ÉVOLUTION DU SOLDE DES REMBOURSEMENTS ENTRE 2003 ET 2012
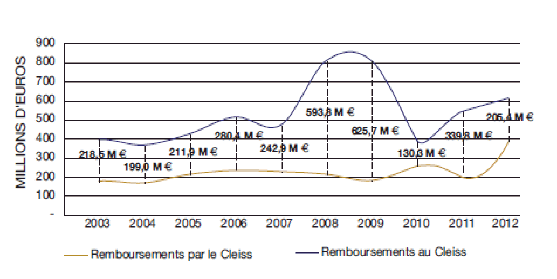
Source : rapport statistique 2012 du CLEISS
À l’examen des statistiques produites par le CLEISS, il apparait que les transferts monétaires liés aux soins transfrontaliers dans l’Union européenne sont favorables à la France. En effet, depuis 2003, le solde entre les remboursements versés par le CLEISS aux autres États membres et ceux perçus par le CLEISS est positif. De même, les créances de la France en matière de soins transfrontaliers ont toujours été supérieures à ses dettes.
La France est donc bien un pays attractif en matière de soins de santé : le développent des soins transfrontaliers au sein de l’espace européen bénéficie à nos comptes sociaux.
Le présent projet de loi permet de parachever l’inscription de la France dans le cadre de l’Europe de la santé.
D’une part, son article premier étend l’obligation d’assurance en responsabilité civile professionnelle aux actes effectués par des ostéopathes et chiropracteurs. Des sanctions sont prévues par l’article 2 en cas de non-respect de cette exigence. Il s’agit de permettre aux patients des ostéopathes et des chiropracteurs de bénéficier de garanties équivalentes à celles existant pour les soins prodigués par des professionnels de santé.
D’autre part, l’article 7 du projet de loi harmonise le contenu des prescriptions transfrontalières en matière de médicaments biologiques. La directive n° 2011/24/UE prévoit en effet que les États membres reconnaissent la validité des prescriptions médicales établies dans d’autres États membres, si elles concernent des médicaments autorisés sur leur territoire. Ces mesures doivent permettre de faciliter la vérification de l’authenticité des prescriptions par les professionnels de santé.
Les progrès de la coopération en matière de santé
Les États membres sont amenés à coopérer pour faciliter la mise en œuvre de la directive. Ils doivent soutenir la création de réseaux européens de référence des prestataires de soins de santé dont l’objet est de contribuer à favoriser la mobilité de l'expertise en Europe.
Les États membres sont également encouragés à collaborer dans le traitement des maladies rares grâce au développement de moyens de diagnostic et de traitement. La base Orphanet et les réseaux européens peuvent être utilisés dans cette optique.
La directive prévoit la mise en place d’un réseau des autorités nationales responsables de la «santé en ligne» en vue de renforcer la continuité des soins et de garantir l’accès à des soins de santé de qualité.
Enfin, la création d'un réseau des autorités ou organes responsables de l’évaluation des technologies de la santé va faciliter la coopération entre les États membres.
Un médicament se définit comme « toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l'homme ou chez l'animal ou pouvant leur être administrée, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique » (article L. 5111-1 du code de la santé publique).
Les médicaments sont de plus en plus couramment l’objet de falsifications donnant lieu à un trafic à l’échelle mondiale. Divers instruments ont été développés pour lutter contre ce phénomène. Le projet de loi vise à les consolider.
La directive 2011/62/UE du 8 juin 2011 définit le médicament falsifié comme « tout médicament comportant une fausse présentation d’au moins l’une des caractéristiques suivantes :
– son identité, y compris de son emballage et de son étiquetage, de sa dénomination ou de sa composition s’agissant de n’importe lequel de ses composants, y compris les excipients, et du dosage de ces composants;
– sa source, y compris de son fabricant, de son pays de fabrication, de son pays d’origine ou du titulaire de son autorisation de mise sur le marché ;
– son historique, y compris des enregistrements et des documents relatifs aux circuits de distribution utilisés. »
On distingue trois catégories de médicaments falsifiés : les médicaments sous standards, les médicaments non-conformes et les faux médicaments. Les médicaments sous-standards sont des médicaments authentiques, produits par les fabricants autorisés par l’autorité nationale de réglementation mais qui ne respectent pas les standards et spécifications de qualité nécessaires pour la commercialisation. Les médicaments non-conformes s’écartent quant à eux des spécifications requises par le dossier d’enregistrement et/ou des bonnes pratiques de fabrication en vigueur dans le pays d’enregistrement. Enfin, les faux médicaments ne sont pas ce qu’ils prétendent être et sont destinés à tromper le consommateur.
Un médicament falsifié peut ainsi contenir les bons ou les mauvais composants, ne pas contenir de principe actif ou seulement en quantité inappropriée ou encore avoir un conditionnement trompeur. Le produit peut-être toxique en lui-même ou bien la faiblesse du dosage en principe actif peut provoquer une résistance aux principes actifs du médicament non contrefait.
On relève des cas de falsification pour toutes les classes thérapeutiques de médicaments.
Le trafic de médicaments falsifiés est un phénomène d’autant plus préoccupant pour la santé publique qu’il est en plein essor. Tous les indicateurs attestent d’une forte croissance de cette activité : nombre de victimes, fréquence des saisies en douane, variété des produits contrefaits… D’après l’Organisation mondiale de la santé (OMS), le chiffre d’affaire mondial du trafic de médicaments falsifiés serait passé de 45 milliards de dollars en 2006 à 75 milliards en 2010.
Cette évolution ne concerne pas uniquement les pays en développement. Le nombre de médicaments falsifiés interceptés par les douanes européennes a plus que quintuplé entre 2005 et 2010 pour atteindre 3,2 millions de médicaments. En France, le nombre de boîtes de médicaments falsifiés saisies a été multiplié par 53 entre 2005 et 2008.
Différents facteurs ont contribué au développement du trafic de médicaments falsifiés.
Tout d’abord, ce trafic est une activité extrêmement lucrative, qui permet de financer les réseaux criminels. Grâce aux profits dégagés par la contrefaçon de médicaments, les trafiquants disposent souvent de moyens supérieurs à ceux des autorités étatiques.
Par ailleurs, la législation de la plupart des pays en développement ainsi que de certains pays développés favorise le trafic de médicaments. De fait, un faible taux de remboursement des médicaments contribue à inciter la population à se procurer sciemment ou non des médicaments falsifiés, moins coûteux. La réglementation relative à la traçabilité des médicaments est également un facteur déterminant, tout comme la complexité de la chaîne d’approvisionnement légale en médicaments. Plus celle-ci est longue, plus le risque de corruption et de substitution par des médicaments contrefaits est élevé. Les peines applicables aux contrefacteurs de médicaments sont par ailleurs très souvent insuffisantes.
Ces failles sont amplifiées par la mondialisation. La croissance des échanges internationaux n’est pas allée de pair avec une augmentation équivalente des moyens permettant de les contrôler, ni avec la création d’une agence internationale chargée de lutter contre le trafic transfrontière de médicaments. Dans l’Union européenne, le principal risque provient des importations parallèles, qui permettent aux grossistes-répartiteur d’un État de s’approvisionner dans un autre État membre au besoin en modifiant le conditionnement des médicaments. Cette pratique, bien que réglementée, constitue une opportunité supplémentaire pour les trafiquants qui utilisent les emballages d’origine pour écouler des médicaments falsifiés ou corrompent les intermédiaires.
Enfin, Internet est un important vecteur de commercialisation de médicaments falsifiés. Il permet aux contrefacteurs d’accéder à un grand nombre de consommateurs, sans aucun contrôle de la qualité ou de l’origine des produits vendus. D’autre part, Internet permet aux acheteurs d’obtenir illégalement certains médicaments, sans prescription médicale. Si l’OMS estime que 50% des médicaments vendus par le biais des sites Internet sont falsifiés, il apparaît en pratique difficile de remonter les filières de contrefacteurs du fait de la courte durée de vie des sites Internet et de l’hétérogénéité des législations nationales.
À l’échelle internationale, INTERPOL coordonne les opérations « Pangéa » organisées chaque année contre la vente sur Internet de médicaments contrefaits. Il s’agit d’opérations faisant intervenir pendant une semaine les douanes, les agences de santé, les polices nationales et les fournisseurs d’accès à Internet de divers pays. La plus grande opération menée, « Pangéa 4 », a rassemblé 81 pays et 165 organismes et permis de saisir 2,4 millions de doses de médicaments en 2011.
L’OMS a pour sa part mis en place le groupe de travail IMPACT afin de lutter contre la contrefaçon de produits médicaux. Ce groupe élabore des recommandations en matière de réglementation, d’investigation, de répression et de communication.
Un cadre de coopération internationale a également été proposé par la convention « Médicrime ». Élaborée par le Conseil de l’Europe mais ouverte à tous, cette convention vise à criminaliser la contrefaçon, la fabrication et la distribution de produits médicaux mis sur le marché sans autorisation ou en violation des normes de sécurité.
L’intervention de l’Union européenne dans la lutte contre le trafic de médicaments se traduit, d’une part, par la conduite d’opérations douanières et, d’autre part, par l’élaboration d’une législation communautaire spécifique.
En 2008, l’Union européenne a mené une action coordonnée, baptisée « MEDI-FAKE », qui a concentré pendant deux mois l’action des douanes des 27 États membres sur l’interception de médicaments contrefaits. Cette opération a conduit à la saisine de 34 millions de médicaments illégaux.
Enfin en 2011, le Parlement européen et le Conseil ont adopté la directive 2011/62/UE visant à prévenir l’introduction de médicaments falsifiés dans la chaîne d’approvisionnement légale. Cette directive impose aux États membres :
- l’apposition de dispositifs de sécurité et de traçabilité sur les emballages des médicaments délivrés sur ordonnance et des médicaments importés ;
- le renforcement du contrôle de la chaîne de distribution des médicaments par de nouvelles responsabilités dévolues aux distributeurs (grossistes et courtiers), par l’inspection des fournisseurs et par l’instauration de systèmes permettant le rappel des médicaments falsifiés;
- la vérification, par les fabricants de médicaments et par les autorités nationales, de la qualité des principes actifs et excipients, quelle que soit leur provenance;
- une harmonisation des législations nationales relatives à la vente en ligne de médicaments (logo commun pour les sites légaux, registre national répertoriant ces sites, sanctions proportionnées et dissuasives).
L’article 5 du projet de loi ratifie l’ordonnance n° 2012-1427 du 19 décembre 2012 qui a transposé la plupart de ces mesures.
La France est relativement épargnée par trafic de médicaments falsifiés. La chaîne de distribution officielle des médicaments est en effet étroitement encadrée. Chacun des acteurs (entreprises pharmaceutiques, grossistes-répartiteurs, pharmaciens d’officine ou d’établissements de santé) a un rôle clairement identifié et est pénalement responsabilisé. Des contrôles sont prévus à chaque étape de la chaîne de distribution par les inspecteurs de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) ou par des pharmaciens inspecteurs des Agences régionales de santé (ARS). Les produits importés par les voies légales sont eux aussi soumis à un contrôle sanitaire.
Par ailleurs, la population est peu incitée à se procurer des médicaments en dehors des réseaux légaux du fait de la qualité de la couverture d’assurance maladie en France et des actions de sensibilisation menées par les autorités.
Des dispositifs visant à éviter l’infiltration par les médicaments falsifiés de la chaîne de distribution légale ont été instaurés. La traçabilité des médicaments a été renforcée depuis l’introduction en 2011 du code DataMatrix pour tous les médicaments. Les industries pharmaceutiques utilisent également des hologrammes et des marqueurs chimiques afin de faciliter la détection des produits contrefaits. Dès 2017, un dispositif permettant de garantir l’intégrité du conditionnement sera mis en place. En outre, le système de rappel et de mise en quarantaine des médicaments falsifiés ou suspects est parfaitement opérationnel.
Les sanctions pénales pour les contrefaçons portant atteinte à la santé et à la sécurité des personnes ont été relevées à 5 ans d’emprisonnement et 500 000euros d’amende par la loi n°2007-1544 du 29 octobre 2007 de lutte contre la contrefaçon (article L. 521-10 du code de la propriété intellectuelle).
Les capacités d’investigations des douanes ont été renforcées dès 2009 par l’adjonction d’équipes de médecins et de pharmaciens inspecteurs. Un plan de lutte contre la contrefaçon des produits de santé a été lancé en 2011 afin notamment de renforcer la coopération opérationnelle entre les services concernés (ANSM, police, gendarmerie, douanes, DGCCRF). Ce plan a abouti à l’élaboration d’un fichier central des vols, détournements et trafics de produits pharmaceutiques.
Des moyens spécifiques ont été mis en place pour lutter la commercialisation frauduleuse par Internet. L’ANSM procède ainsi à l’achat de lots de médicaments vendus en ligne afin de les analyser et de saisir, le cas échéant, les autorités judiciaires compétentes.
La légalisation de la commercialisation des médicaments en accès libre, puis de tous les médicaments non soumis à prescription médicale obligatoire, via les sites Internet des pharmacies physiques rend néanmoins nécessaire la prise de précautions supplémentaires. Dans cette optique, l’article 5 du projet de loi précise et élargit les sanctions applicables en cas de non-respect des exigences posées par l’ordonnance du 19 décembre 2012 ou des bonnes pratiques de dispensation des médicaments par voie électronique définies par l’arrêté du 20 juin 2013.
Au vu de la part des médicaments distribués via Internet, la réglementation de la vente en ligne des médicaments constitue un enjeu important de la lutte contre la contrefaçon.
L’interdiction totale de vente en ligne de médicaments non falsifiés est contraire au droit européen depuis l’arrêt « DocMorris » du 11 décembre 2003 de la Cour de justice de l’Union européenne qui a jugé que les États membres ne peuvent exclure de la vente à distance au public au moyen de services de la société de l’information que les médicaments soumis à prescription.
Certains États-membres, tels l’Allemagne, autorisent la vente en ligne de l’ensemble des médicaments. Ceci semble d’autant plus risqué que les médicaments les plus sujets à la contrefaçon sont précisément les médicaments soumis à prescription médicale obligatoire. Interdire leur vente en ligne permet à l’acheteur qui en trouverait sur un site internet d’identifier immédiatement ce dernier comme le site d’un faussaire.
En outre, les médicaments non soumis à prescription médicale obligatoire, et a fortiori les médicaments présentés devant le comptoir du pharmacien, sont les plus adaptés à l’automédication.
L’article 5 du projet de loi traduit le choix du Gouvernement de n’ouvrir l’exercice de la vente en ligne de médicaments qu’aux seuls pharmaciens déjà titulaires d’une officine. Le site internet de la pharmacie est donc considéré comme le prolongement virtuel d’une officine de pharmacie autorisée et ouverte au public. Le site internet étant adossé à une pharmacie « physique », les patients pourront si nécessaire se rendre dans une officine et échanger avec un pharmacien. Ceci constitue une garantie majeure de sécurité. De même l’interdiction des remises quantitatives évitera la surconsommation de médicaments.
Dans son récent avis du 19 décembre 20135 sur le fonctionnement de la concurrence dans le secteur de la distribution du médicament à usage humain en ville, l’Autorité de la concurrence souligne que « le régime mis en place en France permet d’assurer un niveau de sécurité élevé dans la distribution du médicament par voie électronique et d’écarter les produits falsifiés ». L’Autorité rappelle son soutien de principe à la vente en ligne au motif que « le commerce électronique est un instrument efficace d’animation de la concurrence », mais indique être « consciente des risques en termes de santé publique que le commerce de médicaments peut faire courir aux patients s’il n’est pas encadré. »
Dès lors l’Autorité dit ne pas s’opposer à la conservation d’un lien fort avec le réseau officinal aboutissant à l’interdiction des « pure players », qui interviendraient sur internet sans être adossés à une pharmacie d’officine.
On peut cependant s’interroger sur la capacité des autorités sanitaires et judicaires à contrôler les sites de commercialisation en ligne si ceux-ci venaient à se développer fortement.
En tout état de cause, ce contrôle est rendu encore plus difficile lorsque le site internet ne relève pas directement d’un pharmacien d’officine. Or le choix du maintien du lien entre pharmacie physique et vente en ligne n’est pas aujourd’hui unanime en Europe, certains États-membres autorisant la vente par d’autres intervenants, non adossés à une pharmacie physique.
LES CHOIX DES DIFFÉRENTS ÉTATS-MEMBRES DE L’UNION EUROPÉENNE
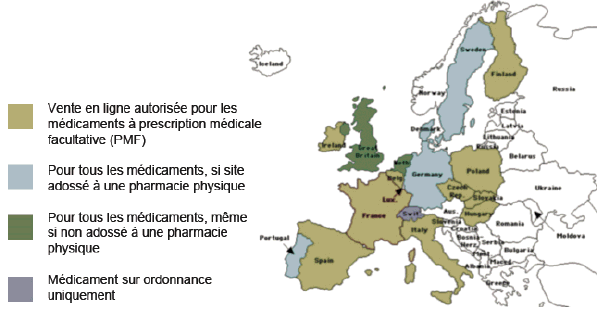
Source : Institut international contre la contrefaçon de médicaments, www.iracm.com
Votre rapporteur estime que l’adaptation au droit de l’Union européenne, ne consiste pas seulement à transposer des normes européennes dans le droit national, au stade ultime de leur élaboration : elle consiste aussi à peser sur le droit européen à venir.
En maintenant le lien entre officine et site internet, la France donne un exemple qui va conforter la position des différents États-membres qui ont choisi cette voie ou s’apprêtent à le faire.
Les études préalables à l’autorisation de mise sur le marché (AMM) ont pour objet d’établir le rapport bénéfices/risques d’un médicament. Elles ne peuvent cependant pas prévoir entièrement les effets indésirables liés à l’utilisation du médicament en conditions réelles.
La pharmacovigilance est définie à l’article L. 5121-22 du code de la santé publique comme « la surveillance, l’évaluation, la prévention et la gestion du risque d’effet indésirable résultant de l’utilisation » des médicaments à usage humain.
Depuis l’instauration du programme de l’OMS pour la pharmacovigilance internationale en 1968, les objectifs de la pharmacovigilance ont évolué, notamment à la suite de scandales sanitaires. L’Union européenne a joué un rôle important dans cette évolution : la directive du 6 novembre 2001 rend ainsi obligatoire la création d’un système de pharmacovigilance pour chacun des États membres de l’Union européenne.
Dans sa version consolidée, l’article 101 de la directive en définit aujourd’hui ainsi les missions :
- recueillir les notifications spontanées des effets indésirables par les professionnels de santé, les patients et les associations agréées de patients ;
- enregistrer et évaluer ces informations ;
- mettre en place des enquêtes ou des études pour analyser les risques, participer à la mise en place et au suivi des plans de gestion des risques ;
- apprécier le profil de sécurité d’emploi du médicament en fonction des données recueillies ;
- prendre des mesures correctives (précautions ou restrictions d’emploi, contre-indications, voire retrait du produit) ;
- diffuser toute information relative à la sécurité d’emploi du médicament, tant au public qu’aux professionnels ;
- participer à la politique de santé publique de lutte contre l’iatrogénie médicamenteuse.
En France, la pharmacovigilance est confiée à l’ANSM et aux centres régionaux de pharmacovigilance (CRPV). Ces derniers recueillent et trient les notifications d’effets indésirables qui leurs sont communiquées, avant de les transmettre à l’ANSM, qui les analyse en une commission nationale de pharmacovigilance. Son directeur général est habilité à prendre les mesures correctives ou préventives nécessaires.
L’organisation est similaire au niveau européen, permettant ainsi une intégration et une complémentarité poussées entre les deux échelons. L’Agence européenne des médicaments (European Medicines Agency, EMA) recueille et valide les notifications transmises par les Etats membres ; en son sein, le comité du médicament à usage humain (CHMP) prend les mesures correctives ou préventives nécessaires, qui s’appliquent de manière harmonisée à l’ensemble des Etats membres.
Une base de données européenne de pharmacovigilance, Eudravigilance, facilite la communication et la collaboration entre les autorités nationales compétentes. Aux termes de l’article R. 5121-156 du code de la santé publique, le directeur de l’ANSM lui « déclare par voie électronique (…) tout effet indésirable grave suspecté, survenu en France, dans un délai de quinze jours à compter de la réception de la déclaration ou de la notification », et « tout effet indésirable non grave suspecté, survenu en France, dans un délai de quatre-vingt-dix jours ». L’article R. 5121-50-1 prévoit en outre que l’Agence « rend accessible, à partir de son site internet, la liste des médicaments, publiée chaque année par l'Agence européenne des médicaments, pour lesquels les autorisations de mise sur le marché ont été refusées, retirées ou suspendues dans l'Union européenne, dont la délivrance a été interdite ou qui ont été retirés du marché »
Peu après qu’une étude indépendante rendue en 2008 à la Commission européenne avait pointé certaines faiblesses du système européen de pharmacovigilance, le scandale du Médiator en apportait la preuve en France.
La réforme initiée à l’échelle européenne par la directive 2010/84/UE du Parlement européen et du Conseil du 15 décembre 2010 relative à la pharmacovigilance a fourni l’occasion au législateur français de réviser en profondeur la pharmacovigilance nationale. Elle a été transposée par la loi n°2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
La directive élargit la définition de l’effet indésirable, qui désigne désormais toute réaction nocive et non voulue à un médicament, et recouvre ainsi les effets résultant d’utilisations non-conformes au résumé des caractéristiques du produit, y compris le mésusage et l’abus de médicaments, ainsi que les effets issus d’erreurs médicamenteuses.
La loi du 29 décembre 2011 ajoute quant à elle les situations où le rapport bénéfice-risque du médicament est défavorable malgré des conditions d’emploi autorisées à la liste des motifs autorisant la suspension ou la modification d’AMM, le retrait d’un médicament du marché et l’interdiction de sa délivrance6.
Surtout, la directive 2010/84 reconnaît aux patients le droit de notifier directement les effets indésirables. Le décret n°2011-655 du 10 juin 2011 a donc modifié l’article R. 5121-154 du code de la santé publique qui prévoit désormais la participation des patients mais également des associations de patients qui « concourent à l’exercice de la pharmacovigilance ».
Enfin, l’article L. 5121-25 créé par la loi du 29 décembre 2011 précise que les médecins, chirurgiens-dentistes, sages-femmes et pharmaciens ont désormais l’obligation de notification, l’article 102 de la directive prévoyant que « les États membres peuvent imposer des obligations spécifiques aux médecins, aux pharmaciens et aux autres professionnels de la santé ».
Parmi les initiatives françaises en matière d’incitation à la notification, votre rapporteur relève l’actualisation des bonnes pratiques de pharmacovigilance, devenues opposables par un arrêté du 28 avril 2005, et diffusées depuis aux professionnels de santé pour les sensibiliser à l’importance de la notification spontanée. L’obligation introduite par la loi du 29 décembre 2011 d’assurer un retour d’information aux notificateurs, qui pourront ainsi constater leur utilité, va elle aussi dans ce sens. Enfin, la notice des médicaments intègre désormais une mention invitant les professionnels de santé et les patients à notifier tout effet indésirable suspecté.
La réussite d’un système de pharmacovigilance repose sur sa capacité à être proactif.
Cela suppose d’abord une évaluation continue des médicaments, et ainsi une mobilisation du titulaire d’AMM. Le droit européen fournit les fondements d’une telle coopération, puisqu’il oblige le titulaire d’AMM à « mettre en oeuvre un système de pharmacovigilance ainsi que d'enregistrer, de déclarer et de suivre tout effet indésirable suspecté d'être dû à un médicament (…) dont il a connaissance » : cette disposition figure désormais à l’article L. 5121-24 du code de la santé publique. Chaque laboratoire dispose à cet effet d’un réseau de correspondants constitué de ses propres chercheurs et visiteurs médicaux, ainsi que de nombreux médecins.
Les législateurs français et européen ont approfondi cette mise à contribution des titulaires d’AMM. L’article R. 5121-168 dispose ainsi que tout titulaire d’AMM « est tenu de transmettre par voie électronique à l'Agence européenne des médicaments un rapport périodique actualisé de sécurité contenant toutes les informations relatives aux bénéfices et aux risques liés à ce médicament ou ce produit (…), une évaluation scientifique du rapport entre les bénéfices et les risques liés au médicament (….) [ainsi que] toutes les informations concernant la vente, la prescription et la population exposée au médicament ou au produit ».
En outre, afin que l’agence dispose d’un tableau de bord à jour recensant non seulement les spécialités approuvées mais aussi celles effectivement commercialisées, l’article L. 5124-5 dispose désormais que l’entreprise exploitant un médicament « communique sans délai, les dates de commercialisation de chaque présentation de ce médicament ou produits».
Enfin, depuis le décret n°2012-1244 du 8 novembre 2012, l’ANSM « peut imposer [au titulaire d’AMM] (…) la réalisation d’études de sécurité post-autorisation (…) [et] d’études d’efficacité post-autorisation lorsque l’acquisition de données supplémentaires est nécessaire après la mise sur le marché du médicament » (article R. 5121-36-1). En outre, le titulaire d’AMM doit mettre en œuvre « un système de gestion des risques pour chaque médicament ou chaque produit » (article R. 5121-163).
Le titulaire d’AMM est enfin tenu de notifier immédiatement à la nouvelle agence toute interdiction ou restriction imposée par l’autorité compétente de tout pays dans lequel le médicament est mis sur le marché et « toutes les informations utiles à l'évaluation des risques et des bénéfices liés à l'emploi des produits qu'il utilise » (article R. 5121-174).
À l’article 6, le présent projet de loi apporte une nouvelle amélioration au système de pharmacovigilance en instaurant un obligation de motivation des décisions de suspension ou d’arrêt de commercialisation de médicament auprès de l’ANSM par l’exploitant du médicament ainsi que des obligations d’information motivée de l’ANSM, sur toute action engagée par le titulaire de l’autorisation de mise sur le marché pour solliciter le retrait de l’autorisation de mise sur le marché ou pour ne pas en demander le renouvellement, ou pour suspendre ou arrêter la commercialisation d’un médicament.
La dernière thématique abordée par le présent projet de loi concerne l’adaptation de la législation française aux dispositions du règlement n° 1223/2009 du Parlement européen et du Conseil relatif aux produits cosmétiques, adopté le 30 novembre 2009 et entré en vigueur le 11 juillet 2013.
Il vise à harmoniser de manière exhaustive les règles applicables dans l’Union européenne en matière de produits cosmétiques, définis comme « toute substance ou tout mélange destiné à être mis en contact avec les parties superficielles du corps humain (épiderme, systèmes pileux et capillaire, ongles, lèvres et organes génitaux externes) ou avec les dents et les muqueuses buccales en vue, exclusivement ou principalement, de les nettoyer, de les parfumer, d’en modifier l’aspect, de les protéger, de les maintenir en bon état ou de corriger les odeurs corporelles ».
L’harmonisation ainsi opérée poursuit un double objectif. Il s’agit, d’une part, d’améliorer le fonctionnement du marché intérieur des produits cosmétiques, et, d’autre part, d’assurer un niveau élevé de protection de la santé humaine. Les bénéfices qui en découlent relèvent donc à la fois de l’activité économique et de la santé.
D’un point de vue économique, l’harmonisation des règles relatives aux produits cosmétiques présente trois intérêts principaux.
• L’harmonisation favorise le développement du libre-échange des produits cosmétiques à l’intérieur de l’Union européenne.
Le règlement européen unifie le droit applicable en matière de produits cosmétiques dans toute l’Union européenne. Cet instrument juridique d’application directe permet en effet d’imposer exactement les mêmes règles à chacun des vingt-huit États membres de l’UE, sans possibilité de transposition divergente.
Il s’agit là d’une avancée importante par rapport à la norme précédemment applicable, la directive 76/768/CEE du 27 juillet 1976, dont la transposition variable avait donné lieu à des différences, parfois significatives, quant aux modalités de commercialisation des produits cosmétiques.
Par exemple, avant la mise sur le marché d’un produit, il ne fallait réaliser aucune démarche spécifique en France mais procéder à une notification en Espagne et déclarer les ingrédients entrant dans la composition en Italie. L’hétérogénéité des règles applicables selon le territoire constituait bien évidemment une entrave à l’exportation des produits cosmétiques.
En outre, la sécurité juridique des industriels du secteur cosmétique était menacée par les perpétuelles modifications apportées à cette directive, révisée à 73 reprises entre 1979 et 2012.
Par ailleurs, les dates d’entrée en vigueur des dispositions de la directive étaient différentes selon les pays. Une même règle pouvait être transposée rapidement dans un Etat membre mais n’être appliquée que plusieurs années après chez ses voisins, ce qui nuisait à l’harmonisation, déjà limitée, des législations. Le règlement permet de répondre à cette difficulté en garantissant la mise en œuvre de ses dispositions à la même date dans l’ensemble de l’Union.
La levée de ces freins à la libre circulation des produits cosmétiques devrait bénéficier à la France, leader mondial dans ce secteur. L’enjeu est de fait considérable pour notre pays dont les entreprises de cosmétiques exportent les deux tiers de leur production et constituent le troisième contributeur de la balance commerciale (pour 7 à 8 milliards d’euros). L’unification de la réglementation au niveau européen en matière de produits cosmétiques profiterait d’ailleurs tant aux grands groupes qu’aux PME, dont le développement est basé sur l’exportation, notamment grâce à la réduction des coûts qu’elle engendre.
• L’harmonisation européenne est en effet source d’une diminution des coûts supportés par les entreprises du secteur cosmétique.
Les coûts d’exportation sont allégés par l’instauration d’une réglementation unique sur tout le territoire de l’Union européenne. De plus, les charges tendent à se resserrer grâce à la simplification réglementaire opérée par la réforme. La substitution d’un seul règlement aux plus de 3 500 pages de réglementations nationales antérieures permet d’accroître la lisibilité du droit applicable, encore renforcée par les clarifications auxquelles le règlement s’est astreint.
Le règlement s’est efforcé de rationaliser la terminologie devenue quelque peu incohérente au fil des révisions successives de la directive. Outre la définition des produits cosmétiques, le règlement a notamment posé une définition commune des nanomatériaux, des effets indésirables ou encore des notions de fabricant, de distributeur et de personne responsable. L’une des vertus de ce texte est d’énoncer très précisément les responsabilités respectives des différents acteurs intervenant dans la chaine d’approvisionnement en produits cosmétiques.
Cependant, le principal vecteur d’économie de la réforme est la mise en place par l’article 13 du règlement d’une procédure centralisée de notification à la Commission européenne préalablement à la mise sur le marché d’un produit.
Autrefois laissée à la discrétion des États membres, cette nouvelle procédure enjoint à la personne responsable du produit de transmettre à la Commission européenne une liste d’informations relatives au produit, à sa composition et à la personne responsable. Cette notification s’effectue par le biais d’une interface électronique (le Portail de Notification des Produits Cosmétiques (CPNP). Ces informations sont ensuite mises à disposition des autorités nationales compétentes et des centres anti-poison, par la Commission européenne. En France, l’autorité compétente en matière de produits cosmétiques est aujourd’hui l’ANSM.
La Commission européenne estime que la simplification de la procédure de notification pour les nouveaux produits cosmétiques devrait conduire à diviser par deux les frais administratifs des entreprises de ce secteur.
• Les nouvelles règles harmonisées améliorent l’information du consommateur
Condition sine qua non d’un marché concurrentiel, l’information des consommateurs dans le domaine des produits cosmétiques est renforcée par le règlement.
En premier lieu, l’article 19 du règlement définit les exigences en matière d’étiquetage des produits cosmétiques. Le récipient et l’emballage du produit doivent porter en caractères indélébiles, facilement lisibles et visibles les informations suivantes : nom ou raison sociale et adresse de la personne responsable du produit, pays d’origine des produits importés, poids ou volume du contenu au moment du conditionnement, date de durabilité minimale du produit, précautions particulières d’emploi, numéro de lot de fabrication ou référence permettant l’identification du produit, et la liste des ingrédients. La langue utilisée pour présenter ces informations est déterminé par l’État membre où le produit est mis à disposition de l’utilisateur final.
D’autre part, le règlement vise à protéger les consommateurs des allégations trompeuses concernant notamment l’efficacité des produits cosmétiques. L’article 20 dispose ainsi que le texte, les dénominations, les marques, les images ou les autres signes figuratifs ne peuvent être utilisés pour attribuer à ces produits des caractéristiques ou des fonctions qu’ils n’ont pas. La Commission européenne a défini, avec les États membres, un plan d’action relatif aux allégations utilisées ainsi que les priorités permettant de déterminer les critères communs justifiant l’utilisation d’une allégation. La liste de ces critères communs, arrêtée par la Commission européenne, est entrée en vigueur en juillet 2013.
Enfin, le règlement facilite l’accès du public aux informations relatives à la formule qualitative du produit cosmétique, à la quantité de substances dangereuses éventuellement présentes et aux effets indésirables potentiels. L’article 21 du règlement exige en effet que ces informations soient rendues facilement accessibles au public, par des moyens appropriés.
La suppression du « visa PP »
Afin d’alléger davantage les charges administratives pesant sur les industriels du secteur cosmétique, l’article 3 du présent projet de loi propose de supprimer le contrôle a priori exercé par l’ANSM sur la publicité relative aux produits, autres que les médicaments, présentés comme bénéfiques pour la santé.
Cette autorisation dénommée « visa PP », actuellement délivrée par l’ANSM après examen de la véracité des allégations publicitaires, est devenue obsolète depuis l’entrée en vigueur du plan d’action européen sur les allégations portant sur les produits cosmétiques et des critères communs justifiant l’utilisation de ces allégations.
Ces nouveaux principes ont encore été précisés par la Charte Publicité et Communication commerciale, adoptée par les organisations professionnelles du secteur dans le cadre de « Cosmetics Europe », ainsi que par la recommandation « Hygiène et beauté » de l’autorité de régulation professionnelle de la publicité (ARPP), adoptée en octobre 2013.
Le règlement réduit donc certaines charges incombant aux entreprises du secteur cosmétique mais il apporte simultanément des améliorations significatives d’un point de vue sanitaire, permettant ainsi de renforcer la sécurité des produits cosmétiques commercialisés dans l’Union européenne.
• Les exigences de sécurité et de traçabilité des produits
La mise en œuvre du règlement européen se traduit par un accroissement des exigences européennes relatives à la sécurité et à la traçabilité des produits cosmétiques. Ceux-ci ne peuvent être mis sur le marché que s’ils satisfont aux normes définies par le règlement dont la plupart concourent à assurer un niveau élevé de protection de la santé humaine.
La conformité à ces règles est garantie par la personne responsable, identifiée pour chaque produit cosmétique. Cette personne, physique ou morale, est en règle générale le fabricant du produit ou son mandataire dans l’UE. Il s’agit toutefois de l’importateur quand le produit est importé dans l’UE, et du distributeur lorsque celui-ci met un produit sur le marché sous son nom ou sa marque, ou bien modifie significativement un produit déjà commercialisé. La personne responsable, en tant que garante de la sécurité du produit, se voit reconnaître diverses obligations.
Elle doit notamment assurer l’évaluation de la sécurité du produit, à partir des exigences minimales définies par le règlement. Ainsi, la personne responsable doit veiller à ce que l’évaluation soit menée sur la base des informations appropriées, en prenant en compte l’usage auquel le produit est destiné et l’exposition systémique attendue aux différents ingrédients de la formulation finale. Toutes les sources existantes doivent être prises en considération, et le rapport de sécurité résultant de cette évaluation doit être actualisé après la mise sur le marché du produit. Enfin, l’évaluation de la sécurité doit être effectuée par une personne titulaire d’un diplôme sanctionnant une formation universitaire en pharmacie, en toxicologie, en médecine ou dans une discipline analogue.
L’exigence de notification à la Commission européenne par la personne responsable préalablement à la mise sur le marché participe également à la protection de la santé humaine, tout comme l’obligation de respect des bonnes pratiques de fabrication harmonisées au niveau communautaire.
Afin de garantir la traçabilité du produit, la personne responsable doit en outre être en mesure d’identifier les distributeurs qu’elle approvisionne en produits cosmétiques, à la demande d’une autorité compétente.
Enfin, en cas de non-conformité d’un produit mis sur le marché, la personne responsable doit informer l’autorité nationale compétente et prendre toutes les mesures nécessaires, y compris des actions correctives de mise en conformité, de retrait ou de rappel du produit.
Le règlement confie des responsabilités particulières aux distributeurs de produits cosmétiques. D’une part, en matière de sécurité, les distributeurs doivent :
- attendre que les mesures nécessaires aient été prises avant de mettre à disposition sur le marché un produit cosmétique qu’ils soupçonnent de ne pas être conforme aux exigences du règlement ;
- s’assurer que les mesures correctives nécessaires ont été prises, s’ils ont des raisons de croire qu’un produit cosmétique qu’ils ont déjà mis à disposition sur le marché n’est pas conforme au règlement ;
- informer immédiatement la personne responsable et les autorités nationales compétentes si le produit concerné présente un risque pour la santé humaine ;
- coopérer avec les autorités compétentes à leur demande afin d’éliminer les risques concernant les produits qu’ils ont mis sur le marché ;
- s’assurer que les conditions de stockage et de transport des produits qui se trouvent sous leur responsabilité ne compromettent pas leur conformité aux exigences du règlement.
D’autre part, en matière de traçabilité des produits cosmétiques, les distributeurs sont tenus, lorsqu’une autorité compétente leur en fait la demande, d’identifier le distributeur ou la personne responsable qui leur a fourni un produit cosmétique ainsi que les distributeurs qu’ils ont eux-mêmes approvisionnés.
Enfin, le règlement s’efforce de garantir la sécurité des produits cosmétiques en restreignant l’utilisation de certaines substances.
Les annexes II et III du règlement établissement la liste des substances dont l’utilisation dans les produits cosmétiques est soit restreinte, soit interdite. De plus, les fabricants ne peuvent utiliser que les colorants, les agents conservateurs et les filtres ultraviolets qui sont explicitement autorisés par les annexes IV à VI.
L’utilisation des substances classées comme cancérogènes, mutagènes ou toxiques pour la reproduction (CMR) est également prohibée, sauf dans des cas exceptionnels.
Une attention particulière est portée à l’utilisation de nanomatériaux, définis comme « un matériau insoluble ou bio-persistant, fabriqué intentionnellement et se caractérisant par une ou plusieurs dimensions externes, ou une structure interne, sur une échelle de 1 à 100 nm ».
L’article 16 du règlement instaure pour tous les produits cosmétiques contenant des nanomatériaux une procédure de notification spécifique à la Commission européenne, six mois avant leur mise sur le marché. Cette notification, effectuée par la personne responsable par voie électronique depuis le 11 janvier 20137, contient des informations relatives à la quantité de nanomatériau utilisé et à ses propriétés.
En cas de doute sur la sécurité d’un nanomatériau, la Commission peut saisir le Comité scientifique pour la sécurité des consommateurs (CSSC) qui dispose alors de six mois pour rendre son avis. À partir de ces informations, la Commission peut réviser les listes des produits dont l’utilisation est limitée ou interdite dans la composition de produits cosmétiques.
• La surveillance par les autorités nationales
Le règlement renforce la surveillance exercée par les autorités nationales sur le marché des produits cosmétiques.
L’article 22 du règlement pose ainsi le principe du contrôle par les États membres du respect des exigences s’appliquant aux produits cosmétiques et notamment de celles relatives à la composition, à la fabrication et à la sécurité du produit. Cette surveillance doit se traduire par des contrôles des produits mis à disposition sur leur territoire mais également des différents opérateurs économiques qui y interviennent. Les autorités nationales compétentes effectuent ce contrôle soit en consultant le dossier d’information conservé par la personne responsable, soit en menant des vérifications, physiques et en laboratoire, sur des échantillons prélevés au préalable.
Lorsque ce contrôle aboutit à une constatation de non-conformité du produit cosmétique, l’autorité nationale compétente exige de la personne responsable qu’elle prenne les mesures appropriées pour y remédier ou, à défaut, pour retirer le produit du marché et le rappeler. Si la personne responsable manque à ses obligations, l’autorité nationale peut arrêter elle-même les dispositions qui s’imposent. L’autorité nationale agit également directement lorsqu’une action immédiate est nécessaire en cas de risque grave pour la santé humaine, mais doit alors en informer la Commission et les autorités compétentes des autres États membres.
• Les progrès de la cosmétovigilance
Le règlement s’attache à définir des règles communes pour la déclaration des effets indésirables liés à l’utilisation d’un produit cosmétique auprès des autorités nationales compétentes. Le système de cosmétovigilance ainsi instauré repose sur la notification des effets indésirables et sur la mise en réseau des autorités nationales.
La notification des effets indésirables graves résultant de l’utilisation d’un produit cosmétique est désormais régie par l’article 23 du règlement. Il s’agit d’imposer à la personne responsable et aux distributeurs de notifier sans délai les effets indésirables graves à l’autorité compétente de l’État membre où ces effets ont été constatés. Cette notification doit comporter la liste de tous les effets indésirables graves dont ils ont connaissance ou dont on peut raisonnablement s’attendre à ce qu’ils aient connaissance ainsi que le nom du produit cosmétique afin de permettre son identification. La personne responsable et les distributeurs doivent également indiquer les éventuelles mesures correctives qui ont d’ores et déjà été prises.
La mise en réseau des autorités nationales compétentes en matière de cosmétovigilance consiste en la transmission des informations reçues concernant les effets indésirables graves qui leur sont notifiés. L’autorité d’un État membre qui a connaissance d’un effet indésirable grave est en effet tenue de transmettre le contenu de la notification qui lui a été adressée aux autorités compétentes de tous les autres États membres.
La réflexion en cours sur l’autorité compétente en matière de cosmétovigilance
Introduit par la loi du 9 août 2004 relative à la politique de santé publique, le système français de cosmétovigilance relève actuellement de la compétence de l’ANSM.
L’article 3 du projet de loi maintient la compétence de l’ANSM en matière de produits cosmétiques comme en matière de tatouage.
La question du transfert de cette compétence à l’Agence nationale chargée de la sécurité sanitaire de l'alimentation, de l'environnement et du travail (ANSES) mérite cependant d’être posée.
Comme le remarque le Docteur Jean-Yves Grall, directeur général de la santé, dans le rapport de mission de juillet 2013 sur la « Réorganisation des vigilances sanitaires », au sein du périmètre de surveillance de l’ANSM « coexistent des vigilances majeures (pharmacovigilance, hémovigilance, matériovigilance) et des vigilances dont l’activité et l’enjeu de sécurité sanitaire sont plus marginaux (cosmétovigilance, produits de tatouage,…) » : ces deux dernières catégories comptent par exemple, en 2011, pour 187 déclarations d’effets indésirables liées aux produits cosmétiques et moins de 10 liées aux produits de tatouage contre près de 60 000 dans le cadre de la pharmacovigilance, 14 000 dans le cadre de l’hémovigilance ou 11 000 dans le cadre de la matériovigilance.
Les signalements relatifs à la cosmétovigilance ont reculé de 26% entre 2009 et 2012. Le nombre de produits cosmétiques contrôlés, quoique variable selon les années, est inférieur à 200.
La cosmétovigilance et la vigilance des produits de tatouage apparaissent donc comme des activités tout à fait marginales de l’ANSM
En outre, la majeure partie des missions de l’ANSM sont liées à une évaluation du rapport risques/bénéfices des produits de santé qui conditionne la délivrance des autorisations de mise sur le marché et des autorisations temporaires d’utilisations. Cette approche est inopérante en matière de produits cosmétiques ou de tatouage, sans effet bénéfique pour la santé, pour lesquels les contrôles consistent généralement à vérifier des concentrations de substances.
La compétence en matière de produits cosmétiques et de tatouage pourrait donc utilement être transférée à l’ANSES, chargée de l’évaluation des risques dans les domaines de l’environnement, du travail et de l’alimentation. L’ANSES est d’ores et déjà habilitée à proposer aux pouvoirs publics toute mesure visant à préserver la santé publique et participe aux travaux des instances européennes et internationales sur les thématiques qui la concernent.
Certaines missions exercées par l’ANSES se rapprochent d’ailleurs de ces vigilances spécifiques, par exemple la nutrivigilance, vigilance sanitaire relative aux compléments alimentaires, aux aliments et boissons enrichis en substances à but nutritionnel ou physiologique, aux nouveaux aliments et aux produits destinés à l’alimentation de populations particulières.
Forte de ses onze laboratoires, de ses 1 350 agents et des 800 experts extérieurs qu’elle mobilise à travers des collectifs d’experts, l’ANSES semble être en mesure d’assumer la vigilance relative aux produits cosmétiques et aux produits de tatouage.
La mesure de l’imputabilité des effets indésirables devrait se développer sous l’effet de cette réforme. En France, l’appréciation par l’ANSM de l’imputabilité d’un effet indésirable à un produit cosmétique devra désormais être rendue publique. Les États membres ne disposant pas des moyens leur permettant d’évaluer efficacement l’imputabilité des effets indésirables bénéficieront des analyses des autres pays.
Par ailleurs, le règlement permet aux autorités compétentes de tous les États membres de demander à la personne responsable, en cas de doute sur la sécurité d’une substance, la liste de tous les produits cosmétiques placés sous sa responsabilité qui contiennent cette substance, en précisant notamment la concentration présente dans chacun d’eux. En France, l’ANSM dispose d’ores et déjà de cette compétence.
Le renforcement de la cosmétovigilance doit permettre d’améliorer la protection de la santé des consommateurs de produits cosmétiques. De fait, cette information accrue sur les produits après leur mise en circulation facilite l’exercice par les autorités nationales de leurs missions de surveillance du marché et d’analyse de la sécurité des produits.
L’harmonisation des règles relatives aux produits cosmétiques telle qu’elle résulte du règlement européen permet ainsi des avancées considérables d’un point de vue sanitaire. Si les nouvelles obligations qui pèsent sur les entreprises du secteur afin de renforcer la sécurité des produits cosmétiques représentent un coût supplémentaire celui-ci sera largement compensé par les réductions de charges découlant des mesures de simplification administrative.
PRINCIPAUX APPORTS DE LA COMMISSION
Sur proposition du rapporteur, la commission a adopté trente amendements. Outre 19 amendements rédactionnels ou de précision et un amendement rectifiant une erreur de référence, la commission a apporté les modifications suivantes :
Á l’article 1, afin de mieux adapter les contrats d’assurance en cours, la commission a différé au 1er janvier 2015 l’obligation de souscription d'une assurance en responsabilité civile professionnelle par les ostéopathes et chiropracteurs.
Á l’article 3 la commission a réaffirmé le principe de reconnaissance automatique entre États membres des formations des personnes chargées d’évaluer la sécurité des produits cosmétiques. Elle a simplifié la distinction entre les effets indésirables graves des produits cosmétiques, soumis à obligation de déclaration, et les autres effets indésirables, en supprimant une catégorie intermédiaire non prévue par le règlement européen. La commission a également distingué la déclaration des effets indésirables et celle des effets résultant d’un mésusage du produit cosmétique. Enfin, la commission a précisé le délai dans lequel les fabricants et distributeurs de produits cosmétiques doivent déclarer les effets indésirables graves.
En matière de tatouages, afin de mieux mesurer la part des effets indésirables résultant de pratiques illégales, la commission a prévu que la déclaration des effets indésirables sera complétée par une description des conditions dans lesquelles le tatouage a été réalisé.
Á l’article 7, afin de permettre la délivrance des dispositifs médicaux dans les différents États membres, la commission a prévu qu’un décret définira les mentions que doit comporter l'acte de prescription.
Enfin, la commission a prévu l’attribution d’un label « éthique » symbolisé par un pictogramme distinctif, qui sera apposé sur les médicaments dérivés du sang issus de la filière du don gratuit et volontaire.
La Commission examine, en première lecture, sur le rapport de M. Olivier Véran, le présent projet de loi au cours de sa séance du mercredi 11 décembre 2013.
Mme la présidente Catherine Lemorton. Ce texte très technique vise à assurer la transposition de directives et la mise en conformité avec le droit communautaire dans le domaine de la santé. Déposé en août dernier, il est inscrit à l’ordre du jour de notre Assemblée le 19 décembre.
M. Olivier Véran, rapporteur. Ce projet de loi portant diverses dispositions d’adaptation au droit de l’Union européenne en matière de santé vise à remplir l’obligation constitutionnelle d’application du droit communautaire qui découle de l’article 88-1 de la Constitution.
L’adaptation au droit européen implique une retranscription fidèle et précise de dispositions que le législateur national ne peut pas modifier sur le fond. Mais les textes européens confèrent également des marges de manœuvre, comme nous le verrons au cours de cet examen.
Il nous revient de traduire dans le droit national les objectifs fixés par plusieurs directives européennes, ce qui n’est nécessaire que dans la mesure où les dispositions nationales n’y satisfont pas déjà. De manière plus contraignante, il nous revient également d’adapter le droit national à un règlement européen. Contrairement à la directive, le règlement est directement applicable, mais ses dispositions peuvent être reprises à des fins d’accès au droit ; elles peuvent également être complétées, dans la mesure où le règlement l’autorise.
Enfin, lorsque la Commission européenne, gardienne des traités, constate que le droit national contrevient au droit européen ou qu’il existe un retard dans la transposition des directives, elle peut engager une procédure d’infraction pour inviter l’État membre à mettre sa législation en conformité. L’État s’expose alors à de lourdes sanctions financières. Aussi, dans tous les cas de figure, une adaptation aussi rapide et complète que possible est nécessaire.
Les mesures d’adaptation visent tout d’abord à parachever la réalisation de la libre circulation des patients en Europe. Les articles 1er, 2 et 7 complètent la transposition des dispositions de la directive du 9 mars 2011 relative à l’application des droits des patients en matière de soins de santé transfrontaliers.
Ce droit est d’ores et déjà largement effectif en France. L’attractivité de notre système de soins nous conduit d’ailleurs à dégager chaque année des excédents : les montants remboursés aux régimes français au titre des soins reçus en France par des personnes affiliées dans d’autres États membres – 615 millions d’euros en 2012 – sont systématiquement supérieurs aux montants des dépenses de santé remboursées par la France au titre de soins reçus dans les autres États membres par des assurés français – 481 millions d’euros en 2012.
La directive conduit à compléter deux aspects du droit existant.
Elle prévoit tout d’abord que les États membres reconnaissent la validité des prescriptions médicales établies dans d’autres États membres pour les médicaments autorisés sur leur territoire. En conséquence, l’article 7 harmonise le contenu des prescriptions transfrontalières de médicaments biologiques au sens du droit européen, c’est-à-dire les médicaments biologiques au sens du code de la santé publique ainsi que les médicaments biologiques similaires, les médicaments immunologiques, les médicaments dérivés du sang et les médicaments de thérapie innovante. La prescription doit comporter la dénomination commune internationale (DCI) des principes actifs d’une part, le nom de marque de la spécialité pharmaceutique d’autre part.
Cependant, la reconnaissance des prescriptions dans les soins transfrontaliers ne sera pleinement effective qu’à l’entrée en vigueur des dispositions de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé, qui a prévu l’obligation de prescription en dénomination commune internationale pour l’ensemble des spécialités. Au préalable, la Haute Autorité de santé (HAS) doit certifier les logiciels de prescription en DCI. Les décrets d’application devraient paraître prochainement et, conformément à la loi, au plus tard le 1er janvier 2015.
La directive prévoit en outre que les prestations de santé délivrées dans les États membres doivent être couvertes par une assurance en responsabilité ou par une garantie équivalente. L’obligation d’assurance en responsabilité civile professionnelle est pleinement effective en France pour toutes les professions de santé. Cependant, elle ne s’étend pas aux actes effectués par des ostéopathes et chiropracteurs, qui ne sont pas reconnus comme professionnels de santé mais qui entrent dans le champ de la directive, puisqu’ils exercent dans le domaine des soins et que leur activité est réglementée.
L’article 1er instaure cette obligation d’assurance. Les patients bénéficieront ainsi des mêmes garanties que pour les soins prodigués par des professionnels de santé, soit jusqu’à 8 millions d’euros par sinistre et 15 millions d’euros par année. En pratique, ces plafonds sont proches de ceux des contrats de groupe qui couvrent aujourd’hui la plupart des ostéopathes et chiropracteurs ; les primes sont peu élevées car les sinistres sont rares. Mais prévoir cette garantie dans la loi est indispensable : il existe en effet plus de 19 000 ostéopathes non médecins en France et ce nombre va doubler dans les prochaines années en raison de la trop grande facilité avec laquelle, entre 2007 et 2012, des organismes privés de formation ont bénéficié d’un agrément. Les nouveaux arrivants disposent en conséquence de formations et de pratiques cliniques hétérogènes, ce qui pourrait entraîner à l’avenir une hausse des sinistres. À cet égard, je me félicite que la ministre des affaires sociales et de la santé ait engagé la réforme du secteur de l’ostéopathie, qui relève du règlement, en concertation avec les professionnels concernés. Il s’agit de mieux définir une formation de qualité et de durcir les critères d’agrément des instituts de formation. De nouveaux décrets sont attendus pour le printemps prochain.
L’article 2 prévoit les sanctions applicables en cas de manquement à l’obligation d’assurance.
En deuxième lieu, le projet de loi adapte les dispositions du code de la santé publique au règlement européen du 30 novembre 2009 relatif aux produits cosmétiques. C’est l’objet de l’article 3.
Le règlement européen se substitue à une directive du 27 juillet 1976 plusieurs fois remaniée et qui était transposée de manière variable dans les différents États membres.
L’unification des règles applicable dans l’Union représente une simplification administrative importante et remarquée, à laquelle nos entreprises du secteur de la beauté ont intérêt. La France est le leader mondial dans ce domaine. Les deux tiers de notre production sont exportés. Le secteur, troisième contributeur de notre balance commerciale avec un excédent de 7 à 8 milliards d’euros, représente 45 000 emplois directs. La baisse des coûts administratifs de mise sur le marché dans les autres États membres n’est pas seulement favorable pour les grands groupes : elle est cruciale pour les PME et les entreprises de taille intermédiaire (ETI), qui représentent 82 % des producteurs et distributeurs de produits cosmétiques.
Le règlement clarifie les responsabilités des intervenants du secteur en définissant les obligations de la « personne responsable » du produit cosmétique, interlocuteur unique des autorités nationales sur l’ensemble du territoire de l’Union européenne et garant de la conformité du produit. Désormais, la notification des informations concernant le produit, préalable à sa mise sur le marché, est centralisée auprès de la Commission européenne, qui transmet les informations aux autorités des États membres. Cette notification doit précéder de six mois la mise sur le marché si les produits contiennent des nanomatériaux. Au vu de l’extrême précision du règlement, la plupart des dispositions de l’article sont de pure retranscription ou de renvoi aux dispositions spécifiques dudit texte.
Les États conservent néanmoins des marges de manœuvre. Le présent article maintient par exemple l’obligation de déclaration préalable à l’Agence nationale de sécurité du médicament (ANSM) de l’ouverture d’un établissement de fabrication ou de conditionnement de produits cosmétiques.
Le règlement prévoit que la personne responsable du produit doit notifier sans délai à l’autorité nationale compétente tous les effets indésirables graves du produit cosmétique dont elle a connaissance ; en retour, l’autorité nationale compétente doit informer ses homologues européens de tous les effets indésirables graves qui lui sont notifiés. Le présent article complète ce dispositif européen de cosmétovigilance en maintenant l’obligation faite aux professionnels de santé de notifier les effets indésirables graves. S’agissant des autres effets indésirables, la loi prévoit une simple faculté de déclaration pour les producteurs, les professionnels de santé, les utilisateurs professionnels et les consommateurs. Ces signaux multiples contribueront à mieux repérer les effets des substances entrant dans la composition des produits cosmétiques.
Par ailleurs, la réglementation des produits de tatouage est actuellement définie en France par renvoi aux règles applicables aux produits cosmétiques ; or les modifications apportées par le règlement européen ne sauraient être étendues, en tant que telles, aux produits de tatouage. L’article 3 rétablit donc dans le code de la santé publique les règles actuellement applicables aux produits de tatouage. Il n’est pas nécessaire de les modifier entièrement : les intervenants du secteur y sont habitués et les coûts occasionnés par des modifications importantes ne seraient pas compensés par des gains tirés de l’harmonisation au plan européen, qui n’existe pas encore.
En matière de médicaments, les articles 5 et 6 achèvent la transposition des objectifs fixés par deux directives qui ont modifié la directive du 6 novembre 2011 instituant le code communautaire relatif aux médicaments à usage humain.
L’article 5 transpose également la directive du 8 juin 2011, relative à la prévention de l’introduction de médicaments falsifiés dans la chaîne d’approvisionnement, en ratifiant l’ordonnance du 19 décembre 2012 prise sur le fondement de l’article 38 de la loi du 29 décembre 2011.
Aux termes de cette ordonnance, un médicament falsifié est un médicament qui comporte une fausse présentation des éléments entrant dans sa composition, ou de son fabricant, ou de l’historique de sa chaîne de distribution. La lutte contre la falsification des matières premières des médicaments est renforcée tant pour les substances actives que pour les excipients : une autorisation de l’ANSM est nécessaire pour toute activité de fabrication ou d’importation. En effet, 80 % de ces matières premières proviennent de pays tiers à l’Union européenne, en particulier d’Inde et de Chine. Les fabricants de médicaments devront vérifier l’authenticité des matières premières et se conformer à des obligations d’audit.
L’activité de courtage en médicament est également réglementée : il s’agit d’une activité exclusivement financière, sans manipulation de lots de médicaments, mais le contrôle des mouvements financiers par l’ANSM renforce la traçabilité des produits et permet de repérer les montages frauduleux liés aux activités des faussaires.
Enfin, la directive a fait le choix d’étendre à l’ensemble des États membres une offre légale de médicaments sur Internet. Au motif de la lutte contre la fraude, dont l’Internet est le principal vecteur, la directive intervient donc dans le domaine de la vente au détail, auparavant réservé aux seuls États membres. Les législateurs nationaux pourraient y voir une atteinte au principe de subsidiarité et envisager de saisir la Cour de justice de l’Union européenne, mais ce serait en vain : la directive se fonde précisément sur l’arrêt dit « DocMorris » du 11 décembre 2003, par lequel la Cour juge que les libertés garanties par les traités ne permettent pas aux États membres d’exclure de la vente en ligne d’autres médicaments que ceux qui sont soumis à prescription.
Je tiens à souligner que ni la Cour ni la directive ne remettent en cause le droit, pour l’État membre, de prévoir le monopole de la délivrance des médicaments par des pharmaciens d’officine.
L’ordonnance a donc autorisé et encadré l’activité de vente en ligne de médicaments. Celle-ci n’est possible qu’à partir du site Internet d’une officine et le site doit être autorisé par le directeur général de l’Agence régionale de santé.
En outre, l’arrêté du 20 juin 2013 relatif aux bonnes pratiques de dispensation des médicaments par voie électronique encadre strictement les modalités de vente afin que le site Internet soit bel et bien le prolongement virtuel d’une officine de pharmacie : la préparation des commandes ne peut se faire qu’au sein de l’officine, dans un espace prévu à cet effet ; la sous-traitance à un tiers est interdite, tout comme les liens hypertexte vers les sites des entreprises pharmaceutiques.
L’ordonnance prévoyait de restreindre la vente en ligne aux seuls médicaments pouvant être présentés en accès direct au public en officine, soit environ 450 médicaments dits « de prescription officinale », qui sont les plus adaptés à l’automédication. Mais cette disposition a été abrogée le 17 juillet 2013 par une décision du Conseil d’État, au motif que le droit européen ne distingue, en vue de l’autorisation de mise sur le marché (AMM), que deux catégories de médicaments : les médicaments soumis à prescription médicale et ceux qui n’y sont pas soumis.
En conséquence, l’article 5 modifie également l’ordonnance afin de prévoir expressément que la totalité des médicaments non soumis à prescription médicale obligatoire pourront être vendus en ligne. Cette catégorie recouvre près de 4 500 spécialités, soit dix fois plus que celle des médicaments en libre accès. Or, sur les onze spécialités à prescription médicale facultative les plus vendues en officine, plusieurs le sont déjà « devant le comptoir ». La disposition n’élargira pas énormément le champ des médicaments disponibles.
D’après les données récentes provenant d’un échantillon de pharmacies bénéficiant de l’autorisation de vente en ligne, après sept mois d’activité, un site Internet a généré environ 235 commandes pour un chiffre d’affaires de 8 128 euros, ce qui est inférieur à 1 % du chiffre d’affaires global de ces officines et à 2,5 % des ventes d’automédication et de parapharmacie.
Rapportée à l’ensemble des officines, la vente en ligne représente 0,01 % du chiffre d’affaires et concerne à 80 % des produits d’hygiène et cosmétologie, des produits de diététique, des compléments alimentaires et des produits pour bébés. L’activité liée aux médicaments est quasi inexistante. La vente en ligne représente donc un avantage particulièrement minime pour le public au regard de la densité et de la qualité du réseau français des pharmacies d’officine, et nous nous en félicitons.
D’ailleurs, ce faible avantage compense mal la part irréductible de risque occasionnée par l’ouverture de la vente en ligne aux médicaments, même non soumis à prescription médicale obligatoire. Les faussaires, on le sait, n’hésitent pas à ouvrir des sites illégaux pour y vendre des médicaments soumis à prescription médicale falsifiés.
Il reste qu’au vu des exigences du droit européen, l’encadrement rigoureux de la vente en ligne proposé par l’ordonnance et par l’arrêté constitue la meilleure solution. Pour les autres États membres qui s’apprêtent à autoriser la vente en ligne, elle est un exemple à suivre. En tout état de cause, il sera sans doute nécessaire, dans quelques années, de modifier les textes européens.
L’article 6 transpose la directive du 25 octobre 2012 relative à la pharmacovigilance, qui définit les nouvelles obligations des titulaires d’autorisation de mise sur le marché. Actuellement, l’obligation d’information de l’ANSM est restreinte aux cas d’arrêt de la commercialisation d’un médicament dans un autre État que la France. Désormais, le titulaire doit informer l’ANSM de toute action qu’il a engagée pour suspendre la mise sur le marché, retirer le médicament du marché, solliciter le retrait de l’AMM ou ne pas en demander le renouvellement. Surtout, il doit informer l’agence des raisons de son action au regard des motifs figurant à l’article L. 5121-9 du code de la santé publique, c’est-à-dire lorsque le médicament concerné est nocif, lorsque l’effet thérapeutique fait défaut, lorsque le rapport bénéfices-risques n’est pas favorable ou lorsque le médicament n’a pas la composition qualitative et quantitative déclarée.
Cette nouvelle obligation de motivation imposée aux titulaires d’AMM permettra d’améliorer l’évaluation bénéfices-risques des médicaments.
Enfin, l’article 4 vise à mettre un terme à une procédure d’infraction à l’encontre de la France pour « entraves à la commercialisation des lentilles de contact » : la Commission a en effet relevé que si la vente en ligne de lentilles correctrices n’est pas interdite en France, les imprécisions des dispositions du code de la santé publique peuvent constituer un obstacle à la libre prestation de services. Après une mise en garde le 27 juin 2007, un avis motivé a été adressé à la France le 18 septembre 2008.
Le risque juridique s’est précisé en 2010 lorsque la Cour de justice a jugé qu’un règlement hongrois qui n’autorise la commercialisation de lentilles de contact que dans des magasins spécialisés est contraire à la libre circulation des marchandises et des services reconnue à l’article 34 du traité ainsi qu’à la liberté d’accès au commerce électronique consacrée par la directive du 8 juin 2000.
Le motif de santé publique avancé pour justifier l’interdiction, sur le fondement de l’article 36 du traité, ne paraissait pas proportionné à l’objectif recherché puisque des mesures moins contraignantes peuvent offrir les mêmes garanties de sécurité : si un État membre peut exiger que les lentilles de contact soient délivrées par une personne qualifiée qui attire l’attention du client sur les risques et l’invite, le cas échéant, à consulter un médecin ophtalmologiste, ces mesures peuvent être prises à distance, par des moyens interactifs, dans le cadre de la vente en ligne.
Aussi, l’article 4 du projet de loi prévoit expressément le cas de vente en ligne de lentilles oculaires correctrices. Cette vente est encadrée : les prestataires concernés doivent permettre au patient d’obtenir informations et conseils auprès d’un professionnel de santé qualifié, donc un opticien-lunetier, un orthoptiste ou un médecin ophtalmologiste.
Ces dispositions ont parallèlement été introduites par amendement à l’article 17 quater du projet de loi sur la consommation, qui les étend en outre à la vente de verres correcteurs. Le contenu des deux textes sera équivalent, sous réserve d’adopter certains amendements à l’article 17 quater, lors de son examen prochain en séance publique, afin de supprimer certaines dispositions qui n’ont rien à y faire – à l’exemple du prolongement de trois à cinq ans de la durée pendant laquelle l’opticien-lunetier peut adapter la prescription du patient.
Le projet de loi sur la consommation est en deuxième lecture et ses dispositions devraient être promulguées avant celles du texte qui nous est soumis ; afin d’écarter au plus vite tout risque de condamnation de la France, il semble préférable de privilégier cet autre vecteur législatif, tout en adoptant, dans un premier temps, l’article 4 tel qu’il nous est présenté. Celui-ci pourrait ainsi être supprimé en séance publique après le vote de l’article 17 quater.
Mme la présidente Catherine Lemorton. Je regrette évidemment la dérégulation que l’Europe nous impose.
M. Arnaud Richard. Ce texte est important, même s’il l’est moins que le projet de loi de financement de la sécurité sociale (PLFSS) ou celui sur les retraites. Par rapport au Parlement européen, nous exerçons un rôle mineur dans ce processus d’application du droit français. Reste que le vote par la représentation nationale est le seul mode de légitimation démocratique des règles décidées au niveau européen. À cet égard, le groupe UDI estime que le Gouvernement doit honorer ses engagements internationaux.
Le projet de loi a un degré de technicité nous incitant à faire confiance à la ministre compétente pour répondre à cette exigence.
Nous avons néanmoins des interrogations sur la volonté du Gouvernement, notamment du ministère de la santé, de jouer le jeu européen. S’il faut saluer son souhait de réduire les délais de transposition des directives et des textes européens, des efforts supplémentaires doivent être faits pour que celle-ci soit de qualité, respecte les délais exigés par l’Union européenne et ne nous oblige pas à agir dans l’urgence.
Si l’on adopte l’amendement du rapporteur qui reporte au 1er janvier 2015 l’obligation de transcription de l’assurance responsabilité civile professionnelle pour les ostéopathes et les chiropracteurs, c’est un peu plus d’un an de retard que la France accuserait par rapport au délai fixé par la directive.
Par ailleurs, nous demeurons pleinement satisfaits sur le fond de ce projet de loi ainsi que sur les amendements de bon sens présentés par le rapporteur, comme celui proposant l’application la plus fidèle possible du droit au principe de libre prestation du service d’évaluation de la sécurité en matière de produits cosmétiques. Plus généralement, toute proposition contribuant à éviter un conflit d’interprétation entre les normes européennes et nationales nous paraît de bon aloi. Nous resterons cependant vigilants pour que les textes européens bénéficient d’une transposition sereine et de qualité en droit français. J’observe à cet égard que les directives européennes sont de plus en plus précises et pointilleuses.
Dans ces conditions, nous voterons en faveur de ce texte pour ne pas perturber la bonne coordination des institutions européennes et nationales.
Mme Bernadette Laclais. Merci au rapporteur pour la précision de son exposé.
La difficulté de ce type de texte est de conserver une visibilité sur la cohérence de l’ensemble de la réglementation, puisqu’il couvre des questions très différentes. Il ne rend pas compte de la complexité des sujets abordés, des avancées, des consensus qui ont dû être trouvés au niveau européen pour aboutir à ces résultats, ni des difficultés de la transposition. Si on peut le regretter, le groupe SRC votera en faveur du projet de loi, en souhaitant que la prochaine mission d’information sur la simplification législative permette de faire en sorte que ce type de texte suscite davantage d’intérêt.
L’article 1er remet en avant la question du statut des professions de chiropracteur et d’ostéopathe : on peut se réjouir des avancées dans ce domaine compte tenu des enjeux qu’elles recouvrent.
S’agissant de l’article 3, nous n’avons pas de remarque particulière à faire, si ce n’est de souligner que la simplification n’exclut pas le renforcement de la sécurité, comme le montrent les propositions qui nous sont faites.
Sur l’article 4, nous nous rangeons à votre proposition de réexaminer la question à la lumière du débat qui se tiendra lundi prochain sur le projet de loi sur la consommation, sachant que nous sommes nombreux à partager le point de vue exprimé par la présidente de notre commission hier en séance publique sur ce texte.
Concernant l’article 5, nous sommes très attentifs sur les propositions relatives au renforcement de la sécurité de la chaîne d’approvisionnement des médicaments. Nous soulignons en outre les difficultés rappelées par un certain nombre de professionnels ainsi que le poids limité que cet aspect représente dans le chiffre d’affaires des entreprises concernées.
Quant à l’article 6, l’obligation de motivation et d’information pour tout ce qui concerne les suspensions ou arrêts de commercialisation de médicaments nous semble un point positif. Il en est de même de l’harmonisation des normes prévue à l’article 7.
M. Arnaud Robinet. Le Gouvernement est en effet très en retard pour transposer ces directives. La disposition sur les ostéopathes et chiropracteurs aurait dû être transposée avant le 17 octobre, celle sur les cosmétiques avant le 11 juillet, celle concernant l’article 6 avant le 28 octobre dernier.
Par ailleurs, l’article 4 tend à mettre notre réglementation en conformité avec les règles européennes à la suite de la décision de la Cour de justice de l’Union européenne du 2 décembre 2010. Or, si l’article additionnel inséré dans le projet de loi sur la consommation a le même objet, il a été rédigé différemment : les relations interministérielles auraient-elles du mal à être opérationnelles au sein du Gouvernement ? Ce ne sont de fait pas les mêmes articles du code de la santé publique qui sont visés.
Dans le projet de loi sur la consommation, le vendeur doit mettre à la disposition du patient un opticien-lunetier, alors que dans le présent texte, on évoque un professionnel de santé qualifié. En ce qui concerne les sanctions, dans le premier, la méconnaissance des règles entraîne une amende de 10 000 euros, alors qu’ici on renvoie à un article du code de la santé publique prévoyant une amende de 3 750 euros. Il va donc falloir faire un choix.
L’encadrement de la vente en ligne des médicaments ne nous pose pas de problème : elle doit rester très restrictive pour protéger les patients. Nous sommes également d’accord avec la nouvelle obligation faite aux ostéopathes de contracter une assurance professionnelle ; en cas de litige pour une faute, elle permettra une meilleure indemnisation des patients.
L’article 6 renforce en effet les obligations existantes pour les titulaires d’autorisation de mise sur le marché des médicaments en introduisant une obligation de motivation de leur décision de suspension ou d’arrêt de leur commercialisation. Pour mémoire, la loi relative aux médicaments de 2011 avait déjà prévu l’obligation pour les entreprises de déclarer à l’Agence du médicament les prescriptions hors AMM de leurs spécialités, ainsi que tout arrêt de commercialisation de leurs produits dans les autres pays – y compris les pays tiers – et toute restriction ou interdiction prise par une autre autorité compétente. L’ensemble de ces dispositions devrait ainsi permettre, après exploitation de ces données, une meilleure police sanitaire des produits.
Le groupe UMP votera donc ce projet de loi, sous réserve de notre interrogation au sujet de l’article 4.
Mme Dominique Orliac. Merci, monsieur le rapporteur, au nom du groupe RRDP, pour la qualité de votre exposé.
J’aimerais revenir sur un point contenu dans l’article 4. En l’état actuel de la législation, la vente à distance de lentilles de contact correctrices n’est pas explicitement interdite sans être non plus clairement autorisée. Or la jurisprudence européenne a indiqué que l’interdiction de cette vente à distance était contraire au droit communautaire.
L’article 4 prévoit donc à la fois d’affirmer la légalité de celle-ci et d’encadrer cette pratique pour assurer la protection des patients. Toutefois, force est de constater que les conditions de première délivrance ne sont pas précisées par le projet et que leur définition est renvoyée à un décret en Conseil d’État.
Je regrette que l’étude d’impact soit le seul document où l’on puisse trouver une référence au contenu envisagé pour ce décret en Conseil d’État. Si cette étude évoque ce contenu au point nommé « Textes d’application » en parlant bien d’une obligation de prescription médicale en cours de validité pour les patients de moins de seize ans, elle ne précise pas ce qu’il doit en être pour les patients plus âgés. Ceux-ci pourraient donc se faire délivrer des lentilles avec une ordonnance datant par exemple de dix ans.
La difficulté est que cette disposition ne peut être précisée par amendement, mais uniquement dans le projet de décret en Conseil d’État.
Le projet de loi sur la consommation prévoit déjà d’étendre les ordonnances de trois à cinq ans, ce qui posera un problème de santé publique, en particulier pour la prévention des maladies ophtalmologiques. À cet égard, il est surprenant que des dispositions ayant des répercussions non négligeables sur la santé des Français soient introduites dans ce texte et soient examinées par la seule Commission des affaires économiques. Je salue à ce sujet les propos tenus hier par la présidente de notre commission.
Pour le dépistage des principales maladies chroniques ophtalmologiques, souvent asymptomatiques au début, la fréquence de trois ans choisie par le législateur en 2007 est clairement appropriée. On sait que plus d’un tiers des patients se voient diagnostiquer une autre pathologie que celle pour laquelle ils sont venus ou un problème de réfraction.
Par ailleurs, le Gouvernement s’est engagé récemment à soutenir les délégations des ophtalmologues vers les orthoptistes. Les expérimentations en cours dans ce domaine sont à mon avis très prometteuses et règleront certainement une grande part des problèmes, en permettant une meilleure fluidité des rendez-vous. Dans le cadre de ces délégations, il est prévu des renouvellements pour des patients vus depuis moins de cinq ans, mais avec des conditions très strictes à respecter, énoncées par la Haute autorité de santé.
Dans le même esprit, le fait qu’aucune mention ne soit explicitée dans l’article 4 de ce projet de loi risque de créer des problèmes majeurs : la vente de lentilles de contact n’est pas un produit de consommation banal, mais un produit médical qui, s’il est mal utilisé, peut entraîner des risques dangereux pour l’œil, tels que des infections – dont certaines peuvent conduire à la cécité. Il y aurait aujourd’hui environ 600 patients hospitalisés par an pour des problèmes d’abcès de cornée, dus à des lentilles mal adaptées.
Il faut donc s’assurer que les patients ne puissent se faire délivrer des lentilles de contact avec des ordonnances de plus de trois ans, trouver un moyen pour que ce délai soit pris en compte dans le décret en Conseil d’État, mais aussi que la prescription initiale soit respectée et qu’il n’y ait pas de modification des paramètres des lentilles de contact lors de ventes promotionnelles par exemple.
Mme la présidente Catherine Lemorton. Merci à tous ceux qui m’ont soutenu pour mon intervention d’hier soir : le champ de compétence de notre commission doit être respecté.
M. Christian Hutin. Monsieur le rapporteur, vous avez réussi à enrichir ce texte par la clarté de vos explications.
L’article 17 quater du projet de loi sur la consommation prévoit de supprimer la condition de diplôme d’opticien-lunetier pour le directeur ou le gérant d’un établissement d’optique lunetterie. Or on sait qu’un grand nombre de patients demandent à leur médecin généraliste des ordonnances pour faire réaliser des lunettes, sans avoir fait de vérification préalable. Il me semble que ces conditions de diplôme existent en France pour le moment dans chaque magasin d’optique, notamment dans les grands groupes : est-ce bien le cas ? Quels contrôles existent sur la présence de personnes diplômées dans ces magasins d’optique, à l’exemple de ce qui prévaut pour les pharmacies ? Ces contrôles sont-ils les mêmes dans les autres pays d’Europe ?
M. Jean-Pierre Barbier. L’Europe modifie effectivement les procédures de délivrance pour un certain nombre de produits, considérés chez nous comme des produits de santé, inscrits au code de la santé publique. Ce texte va dans le bon sens pour les lentilles correctrices et le médicament, et je remercie le rapporteur pour ces explications. Je vous remercie également, madame la présidente, pour les propos que vous avez tenus hier soir.
Cela étant, je suis inquiet au sujet de ce que j’ai entendu à cette occasion dans l’hémicycle : quand on considère uniquement le prix et la rapidité de la délivrance, je crains qu’on se laisse influencer par les demandes du lobbying des grandes surfaces. Le prix ne peut tout justifier. Si l’on décide ainsi que les tests de grossesse peuvent être vendus en grande surface au motif qu’ils sont déjà sur Internet, on risque de banaliser les produits de santé. Je redoute que ce que nous avons acté aujourd’hui au sein de notre commission soit à nouveau balayé demain par le projet de loi sur la consommation. Nous devons être très vigilants à cet égard.
M. Gérard Bapt. J’ai voté hier soir en faveur de l’amendement de suppression de l’article 17 quater A. On voit bien dans les études cliniques que la plupart des abcès rétiniens se produisent sous lentilles et qu’il existe une utilisation inappropriée de celles-ci en raison d’un manque d’information des patients sur les risques d’infection. S’il n’est pas possible de revenir sur la disposition de vente sur Internet des lentilles de contact, la vente des produits de nettoyage devrait être réservée à des professionnels tels que les pharmaciens ou les opticiens. Ceux-ci pourraient expliquer qu’avant de changer de lentilles, il convient de se laver les mains, ou qu’il ne faut pas les mettre pour aller dans une piscine.
M. le rapporteur. Je m’associe également aux éloges qui vous ont été adressés, madame la présidente, pour votre intervention d’hier. Il est hors de question d’aborder les problématiques de santé dans un cadre strictement économique.
Monsieur Robinet, le règlement communautaire n’a pas attendu ce projet de loi pour être appliqué : il l’est déjà de façon directe depuis juillet dernier. Nous ne sommes donc pas en infraction à cet égard.
S’agissant de l’article 4, un amendement à l’article 17 quater du projet de loi sur la consommation ira dans le sens du présent projet de loi, notamment s’agissant du professionnel de santé qualifié. La seule différence est que la vente en ligne concernera aussi les verres correcteurs. Je rappelle que la vente en ligne de lentilles existe depuis 2005, en dépit de l’absence de réglementation l’autorisant.
Madame Orliac, l’obligation d’ordonnance pour les lentilles de contact n’existe aujourd’hui que pour les mineurs de moins de seize ans : s’il n’y aura pas de modification des conditions de prescription dans le décret, le remboursement n’est possible que sur présentation d’une ordonnance, ce qui peut inciter à en obtenir une. Par ailleurs, ces produits constituent un dispositif médical de classe II a, correspondant à un risque moyen.
Monsieur Hutin, s’agissant des magasins d’optique, conformément à la réglementation européenne, nous allons passer d’un monopole de gestion à un monopole de délivrance : la loi précisera que seul un professionnel pourra délivrer des lentilles de contact.
Article 1er
Responsabilité civile professionnelle des chiropracteurs et des ostéopathes
L’article premier instaure une obligation d’assurance de responsabilité civile professionnelle (RCP) pour les chiropracteurs et les ostéopathes.
Cette mesure vise à mener à terme la transposition de la directive 2011/24/UE du 9 mars 2011 relative à l’application des droits des patients en matière de soins transfrontaliers au sein de l’Union dont l’article 4 prévoit que les États membres s’assurent de l’existence de « systèmes d’assurance responsabilité professionnelle » pour les traitements dispensés sur leur territoire.
Il s’agit de garantir à l’ensemble des citoyens de l’Union européenne faisant usage de leur droit d’accéder à des soins dans un État-membre différent de l’État d’affiliation qu’ils seront effectivement indemnisés en cas de sinistre intervenu à l’occasion de ces soins.
L’obligation d’assurance existe d’ores et déjà en France pour les actes relevant des professions de santé régies par le code de la santé publique : le présent article l’étend aux actes des chiropracteurs et des ostéopathes, qui ne relèvent pas du code de la santé publique.
1. L’absence actuelle d’obligation d’assurance
L’ostéopathie concerne l’ensemble des manipulations manuelles externes ayant pour but de prévenir ou de remédier à des troubles fonctionnels du corps humain. Près de 19 000 professionnels usent légalement du titre d’ostéopathe. Cet effectif pourrait doubler dans les prochaines années en raison de l’augmentation de l’offre de formation, privée, au cours de la dernière décennie.
L’ostéopathie se distingue de la chiropraxie qui recouvre l’ensemble des actes de manipulation et de mobilisation externes ayant pour but de prévenir ou de remédier à des troubles de l’appareil locomoteur du corps humain. On dénombre environ 900 chiropracteurs en exercice.
Ces pratiques ne sont pas reconnues comme relevant d’une profession de santé et les actes ne sont pas remboursés par l’assurance maladie obligatoire.
Dès lors, l’obligation d’assurance pour responsabilité civile professionnelle des professionnels de santé, prévue par l’article L. 1142-2 du code de la santé publique, qui couvre uniquement « leurs activités de prévention, de diagnostic et de soins », n’est pas applicable aux ostéopathes et chiropracteurs.
Ainsi, dans les cas où un professionnel autorisé à faire usage d’un de ces titres est par ailleurs professionnel de santé (kinésithérapeutes par exemple), l’assurance professionnelle RCP prévue par la loi ne couvre pas les actes liés à l’exercice de l’ostéopathie : le professionnel est amené à compléter son contrat d’assurance par des clauses particulières.
Cette distinction établie par le droit national est cependant inopposable à la directive 2011/24 dont l’article 3 définit les professionnels de santé de façon plus large : la notion recouvre non seulement les professionnels de santé au sens du droit français (médecins, infirmiers, pharmaciens, sages-femmes, kinésithérapeutes) mais également toutes les personnes pratiquant, dans le secteur des soins ou de la santé, une profession réglementée c’est-à-dire une « une activité […] dont l’accès, l’exercice ou une des modalités d’exercice est subordonné directement ou indirectement, en vertu de dispositions législatives, réglementaires ou administratives, à la possession de qualifications professionnelles déterminées ».
L’ostéopathie et la chiropraxie relèvent bien des professions réglementées entrant dans le champ d’application de l’article 4 de la directive 2011/24 car l’article 75 de la loi du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé réserve l’usage de ce titre professionnel « aux personnes titulaires d’un diplôme sanctionnant une formation spécifique ».
Cette disposition a été précisée, pour les ostéopathes, par les décrets n° 2007-435 et -437 du 25 mars 2007 relatifs respectivement aux actes et conditions d’exercice de l’ostéopathie et à la formation des ostéopathes et à l’agrément des établissements de formation.
L’exercice de la chiropraxie est régi par le décret n° 2011-32 du 17 janvier 2011 relatif aux actes et aux conditions d’exercice de la chiropraxie et le décret n° 2011-1127 du 20 septembre 2011 relatif à la formation des chiropracteurs et à l’agrément des établissements de formation en chiropraxie.
Aucune obligation légale n’impose par conséquent aux ostéopathes et aux chiropracteurs de souscrire une assurance responsabilité civile. De fait, lorsqu’aucune assurance en RCP n’est souscrite, les victimes d’une faute professionnelle ne peuvent être indemnisées que dans la limite du patrimoine propre du praticien.
À défaut d’obligation légale, on relève néanmoins que la plupart des associations professionnelles de chiropracteurs et d’ostéopathes proposent à leurs membres de souscrire un contrat de groupe comportant une assurance RCP. Ainsi, la grande majorité des personnes exerçant ces activités, qu’il s’agisse de professionnels de santé ou de praticiens à titre exclusif, sont couvertes par une garantie spécifique.
2. L’alignement partiel sur l’assurance des professionnels de santé
Le présent article instaure pour les chiropracteurs et les ostéopathes une obligation d’assurance responsabilité civile proche de celle couvrant les activités de prévention, de diagnostic ou de soins des professionnels de santé.
L’alinéa 1 fait reposer l’engagement de la responsabilité sur l’existence d’une faute commise dans le cadre de l’activité professionnelle. À ce nouveau cas d’engagement de la responsabilité s’ajoute la responsabilité en raison du défaut d’un produit de santé, régie par les dispositions de la loi n° 98-389 du 19 mai 1998 relative à la responsabilité du fait des produits défectueux.
L’alinéa 2 établit l’obligation d’assurance en RCP des ostéopathes et chiropracteurs exerçant à titre libéral : l’assurance doit couvrir les dommages subis par des tiers et résultant d’atteintes à la personne.
L’alinéa 3 offre la possibilité aux contrats d’assurance de fixer des plafonds de garantie, mais ces plafonds ne sauraient être inférieurs à des montants déterminés par un décret pris en Conseil d’État. L’étude d’impact jointe au projet de loi indique que ces plafonds seront les mêmes que ceux des assurances des professionnels de santé, soit 8 millions d’euros par sinistre et 15 millions d’euros par année.
Les représentants des ostéopathes et des chiropracteurs auditionnés par votre rapporteur ont indiqué que ces montants sont proches de ceux prévus par les contrats en vigueurs, mais des modifications devront néanmoins être apportées, occasionnant une hausse modérée des primes d’assurance. En effet certains contrats de groupe prévoient aujourd’hui une mutualisation du montant du plafond annuel à l’échelle de l’ensemble des souscripteurs : ils devront désormais prévoir un plafond annuel par membre du groupe.
L’alinéa 4 rend applicables aux contrats d’assurance civile des ostéopathes et des chiropracteurs les dispositions prévues aux articles L. 251-2 et L. 251-3 du code des assurances. Le contrat d’assurance civile de ces professionnels devrait ainsi garantir :
– les sinistres pour lesquels la première réclamation intervient au cours de la période de validité du contrat ;
– les sinistres résultant d’un fait dommageable survenu pendant la validité du contrat mais pour lesquels la première réclamation intervient dans un délai qui ne peut être inférieur à cinq ans à compter de la résiliation ou de l’expiration du contrat (garantie subséquente) ;
– lorsqu’il s’agit du dernier contrat conclu avant la cessation d’activité ou le décès de l’assuré, les sinistres résultant d’un fait dommageable survenu pendant la validité du contrat mais pour lesquels la première réclamation intervient dans un délai qui ne peut être inférieur à 10 ans à compter de la résiliation ou de l’expiration du contrat.
L’alinéa 5 prévoit l’entrée en vigueur de l’obligation d’assurance en RCP au 1er janvier 2014. Or, à cette date, le projet de loi ne sera pas entré en vigueur. Comme l’article 2 du projet de loi punit le manquement à l’obligation d’assurance institué par le présent article, votre rapporteur considère que le principe de non-rétroactivité de la mesure pénale plus sévère oblige à modifier cette date.
Le présent article aligne donc sur le nouveau régime de RCP des ostéopathes et des chiropracteurs sur celui des professionnels de santé. Néanmoins, le maintien de la spécificité de l’usage des titres emporterait le maintien de certaines différences.
En premier lieu, l’assurance en RCP instaurée pour les ostéopathes et les chiropracteurs ne couvrirait pas les actes commis par leurs salariés, contrairement à celle des professionnels de santé.
En second lieu, la sinistralité connue en matière d’ostéopathie et de chiropraxie étant très faible, et n’ayant en outre jamais atteint les montants de plafonds de garantie envisagés, les ostéopathes et chiropracteurs ne cotiseraient pas au fonds complémentaire d’indemnisation prévu, pour les seuls professionnels de santé, à l’article L. 1142-2 du code de la santé publique. Ce fonds intervient lorsque les dommages sont supérieurs à 8 millions d’euros par sinistre, la victime bénéficiant alors de la prise en charge de la fraction de l’indemnisation dépassant le plafond de garantie fixé par le contrat d’assurance, sans possibilité d’action récursoire contre le professionnel de santé.
Enfin, l’attention de votre rapporteur a été attirée sur l’absence d’indemnisation de la victime dans les cas d’absence de faute, puisque l’intervention de l’Office national d’indemnisation des accidents médicaux (ONIAM) est restreinte à la réparation de sinistres intervenus à l’occasion de la pratique des professionnels de santé énumérés dans la 4e partie du code de la santé publique, au nombre desquels les chiropracteurs ou ostéopathes ne figurent pas.
*
La Commission adopte l’amendement AS12 rédactionnel du rapporteur.
Elle examine ensuite l’amendement AS1 du rapporteur.
M. le rapporteur. Cet amendement propose de reporter au 1er janvier 2015 la date d’entrée en vigueur de l’obligation d’assurance des ostéopathes et chiropracteurs – au lieu de la prévoir le 1er janvier 2014, date à laquelle la loi ne sera pas encore applicable.
La Commission adopte l’amendement.
Puis elle adopte l’article 1ermodifié.
Article 2
Sanction du manquement à l’obligation d’assurance de responsabilité civile professionnelle des chiropracteurs et des ostéopathes
Le présent article définit les sanctions applicables en cas de non-respect des dispositions de l’article premier.
L’alinéa 1 prévoit de sanctionner les ostéopathes et les chiropracteurs exerçant sans assurance responsabilité civile d’une amende de 45 000 euros.
L’alinéa 2 introduit une peine complémentaire d’interdiction d’exercice de l’activité professionnelle ou sociale. Cette interdiction d’exercer pourrait être prononcée soit à titre définitif, soit à titre provisoire, pour une durée maximale de cinq ans.
Il s’agit des mêmes sanctions pénales que celles qui s’appliquent aux professionnels de santé exerçant des activités de prévention, de diagnostic ou de soins en cas d’absence d’assurance responsabilité civile. Dans ce cas, les professionnels de santé s’exposent cependant en outre à des sanctions disciplinaires spécifiques, en vertu de l’article L. 1142-2 du code de la santé publique, qui ne sont pas applicables aux ostéopathes et chiropracteurs.
*
La Commission adopte l’article sans modification.
Article 3
(art. L. 5122-14, L. 5131-1 à L. 5131-11, L. 513-10-2, L. 513-10-3, L. 513-10-4, L. 513-10-5 à L. 513-10-10 [nouveaux], L. 5431-2, L. 5431-5, L. 5431-6, L. 5431-7, L. 5431-8 et L. 5431-9 [nouveaux], L 5437-2, L 5437-3 à L 5437-5 [nouveaux] du code de la santé publique)
Produits cosmétiques et de tatouage
L’article 3 adapte les dispositions du code de la santé publique relatives aux produits cosmétiques en raison de l’entrée en vigueur, le 11 juillet 2013, du règlement européen n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques.
Ce règlement est d’application directe. La mise en cohérence du code de la santé publique avec les dispositions principales de la réglementation européenne vise donc à donner une information claire et cohérente sur le droit applicable. C’est l’objet des sections I, II, V et VI du présent article.
Dans la mesure où la réglementation des produits de tatouage est actuellement définie en France par renvoi aux dispositions relatives aux produits cosmétiques modifiées par l’article 3, les sections III et IV du présent article établissent directement les règles applicables aux produits de tatouage, bien qu’elles ne relèvent pas du champ du règlement européen.
1. Les produits cosmétiques
Le règlement européen se substitue à une directive 76/768/CEE du 27 juillet 1976 ayant le même objet mais plusieurs fois remaniée et qui était transposée de manière variable dans les différents États membres.
Un règlement européen étant d’application directe, ses effets sont simultanés, automatiques et uniformes au sein de l’Union européenne, ce qui simplifie les procédures pour les opérateurs économiques du secteur et constitue un gage supplémentaire de sécurité des produits cosmétiques.
Le règlement européen clarifie les responsabilités des intervenants du secteur en définissant, à l’article 4, les obligations de la « personne responsable » du produit cosmétique, interlocuteur unique des autorités nationales, sur l’ensemble du territoire de l’Union européenne. Elle est la personne qui met le produit cosmétique sur le marché et est chargée d’en garantir la conformité aux obligations établies dans le règlement.
Symétriquement, l’article 34 du règlement prévoit que les États membres désignent une « autorité compétente nationale », garante du respect des règles en matière de produits cosmétiques.
Au nombre des obligations reposant sur les personnes responsables figurent :
– le fait de garantir la conformité du produit cosmétique aux obligations d’évaluation de la sécurité définies par le règlement (article 10),
– la conservation d’un dossier d’information sur le produit (article 11),
– la notification à la Commission européenne avant mise sur le marché, prévue à l’article 13, des informations principales concernant le produit via le CPNP (Cosmetics Products Notification Portal – Portail de Notification des Produits Cosmétiques). Ces informations recouvrent, par exemple, la dénomination des substances classées comme cancérogènes, mutagènes ou toxiques pour la reproduction (CMR) ou la formulation-cadre permettant un traitement médical approprié en cas de trouble. Pour les produits contenant des « nanomatériaux », l’article 16 prévoit que la notification doit intervenir au moins six mois avant la mise sur le marché.
De même l’article 19 du règlement clarifie les obligations en matière d’étiquetage et renforce les obligations incombant au distributeur du produit, défini comme « toute personne physique ou morale faisant partie de la chaîne d’approvisionnement, autre que le fabricant ou l’importateur qui met un produit cosmétique à disposition sur le marché communautaire ». Cette définition inclut tant les personnes qui opèrent dans le commerce de gros que les détaillants qui vendent directement au consommateur, par exemple dans les salons de coiffure ou d’esthétique.
a) De nombreuses mesures de stricte adaptation
Les dispositions du code de la santé publique relatives aux produits cosmétiques sont actuellement définies aux articles L. 5131-1 à L. 5131-11.
Le présent article modifie l’ensemble de ces dispositions.
L’alinéa 2 rectifie, à l’article L. 5131-1, la définition des produits cosmétiques afin de la calquer sur celle qui figure à l’article 2 du règlement européen selon lequel il s’agit de « toute substance ou tout mélange destiné à être mis en contact avec les parties superficielles du corps humain (épiderme, systèmes pileux et capillaire, ongles, lèvres et organes génitaux externes) ou avec les dents et les muqueuses buccales en vue, exclusivement ou principalement, de les nettoyer, de les parfumer, d’en modifier l’aspect, de les protéger, de les maintenir en bon état ou de corriger les odeurs corporelles ».
L’alinéa 7 complète l’article L. 5131-2 en précisant que l’obligation de désigner des personnes qualifiées en charge de l’évaluation de la sécurité des produits cosmétiques avant leur mise sur le marché, est satisfaite si les personnes concernées possèdent les qualifications mentionnées à l’article 10 du règlement européen : il s’agit de titulaires d’un titre sanctionnant une formation universitaire en pharmacie, toxicologie, médecine ou dans une discipline analogue, ou une formation reconnue équivalente par un État membre. Il en résulte un principe de reconnaissance mutuelle des qualifications reconnues comme équivalentes par les différents États membres. Dans la rédaction projetée de l’article L. 5131-2, la formation équivalente devrait figurer sur une liste établie par arrêté : votre rapporteur souligne que l’établissement cette liste ne saurait faire obstacle à la reconnaissance d’autres formations valablement reconnues comme équivalentes dans d’autres États-membres.
L’alinéa 9 modifie l’article L. 5131-3 qui établit l’obligation pour les produits cosmétiques de satisfaire aux dispositions du règlement européen, précédemment décrites.
Conformément à l’article 34 du règlement qui prévoit que « les États membres désignent leurs autorités compétentes nationales », l’alinéa 10, au même article L. 5131-3, désigne l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) comme l’autorité compétente au sens du règlement européen ; ses différentes missions et prérogatives en matière de produits cosmétiques figurent par renvoi aux articles du règlement européen qui les instituent, à savoir :
– la mission de vérification de la conformité de l’étiquetage (paragraphe 5 de l’article 6 du règlement) ;
– le droit d’accès au dossier d’information (paragraphe 3 de l’article 11) ;
– la transmission par la commission européenne des informations provenant de la notification centralisée (paragraphe 5 de l’article 13) ;
– la mission de recevoir les notifications d’effets indésirables graves des produits cosmétiques effectuées dans les États membres et la mission de communiquer ces effets aux autorités responsables des autres États-membres (article 23) ;
– le droit de demander des informations à la personne responsable en cas de doute sérieux quant à la sécurité de toute substance entrant dans la composition des produits cosmétiques (article 24) ;
– le pouvoir de prendre toutes les mesures appropriées, proportionnées à la nature du risque, y compris des actions correctives de mise en conformité du produit cosmétique, de retrait du marché ou de rappel, en cas de constat d’une non-conformité (article 25) ou d’exiger des distributeurs des produits cosmétiques qu’ils prennent les mêmes mesures (article 26) ;
– le pouvoir de prendre toutes les mesures provisoires appropriées pour obtenir le retrait de produits pouvant présenter un risque grave pour la santé humaine, conformément à la « clause de sauvegarde » de l’article 27 du règlement européen ; les mesures prises sont alors immédiatement communiquées à la commission européenne et aux autorités compétentes des autres États membres ;
– l’obligation, dans l’exercice de ses compétences, de se conformer aux bonnes pratiques administratives définies à l’article 28 du règlement, telles que la motivation des décisions et le respect du principe du contradictoire, au demeurant pleinement garanties par les principes fondamentaux du droit administratif français ;
– enfin l’obligation de coopérer avec les autorités compétentes des autres États-membres par échange d’expérience (article 29) ou en matière de vérification du contenu du dossier d’information sur le produit cosmétique de la personne responsable d’un produit (article 30).
L’article L. 5131-3 désigne également comme autorités compétentes le ministre chargé de la consommation ainsi que les agents de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), de la direction générale des douanes et de la direction générale des finances publiques. Ils sont dotés des mêmes compétences à deux exceptions près : contrairement à l’ANSM, ils ne peuvent pas utiliser la clause de sauvegarde ; de même, ils ne reçoivent pas notification des effets indésirables, mais le paragraphe 5 de l’article 23 du règlement européen leur donne le droit d’utiliser ces informations à des fins de surveillance et d’information des consommateurs.
L’alinéa 11 modifie l’article L. 5331-4 : la mention de l’obligation pour les produits cosmétiques mis sur le marché de ne pas nuire à la santé humaine dans des conditions normales d’utilisation est supprimée car elle est rendue inutile par le renvoi direct aux définitions plus précises du règlement, opéré par la nouvelle rédaction de l’article L. 5331-3.
Désormais, l’article L. 5331-4 prévoit que l’ANSM publie les principes de bonnes pratiques des laboratoires en matière de produits cosmétiques. Ces principes sont définis par la directive 2004/10/CE, concernant « le rapprochement des dispositions législatives, réglementaires et administratives relatives à l’application des principes de bonnes pratiques de laboratoires et au contrôle de leur application pour les essais sur les substances chimiques ». Pour les produits cosmétiques, ces principes ont été transposés en droit français, par un arrêté du 10 août 2004. En matière d’inspection et de vérification des bonnes pratiques de laboratoire, l’ANSM dispose à nouveau d’une simple compétence de publication des règles applicables, qui sont définies par une directive 2004/9/CE qui a été transposée en droit français, pour les produits cosmétiques, par la décision de l’ANSM du 15 novembre 2006 prise en application de l’article L. 5311-1 du code de la santé publique. En revanche, l’ANSM est chargée de définir les règles relatives à la délivrance des documents attestant le respect de ces bonnes pratiques.
En conséquence, les dispositions de l’actuel article L. 5131-5, qui ont également trait aux bonnes pratiques de fabrication, sont supprimées.
Enfin, d’autres dispositions actuelles sont abrogées en raison de l’application directe de prescriptions du règlement européen de portée équivalente voire strictement identique. Ainsi des dispositions en matière d’expérimentation animale à l’article 18 du règlement, privilégiant le recours à des méthodes alternatives, qui ont le même objet que les dispositions actuelles de l’article L. 5131-7-2, abrogé en conséquence. De même, l’article L. 5131-7-1 est abrogé au vu de l’article 21 du règlement qui définit l’obligation, pour la personne responsable, de permettre l’accès du public aux informations déterminantes sur les ingrédients du produit cosmétique et sur leurs effets indésirables.
Enfin, le V du présent article, à l’alinéa 94, abroge l’article L. 5131-7 du code de la santé publique qui institue un contrôle préalable de l’ANSM, couramment qualifié de « visa PP », de la publicité en faveur de produits autres que les médicaments présentés comme bénéfiques pour la santé. Il s’agit des produits alléguant qu’ils favorisent une modification de l’état physique ou physiologique. Conformément à l’article 20 du règlement européen, s’y substitue un « plan d’action relatif aux allégations concernant le produit » cosmétique. Cette réglementation est intervenue sous la forme d’une « Charte Publicité et Communication commerciale » adoptée par les organisations professionnelles du secteur dans le cadre de « Cosmetics Europe », complétée, en France, par une recommandation « Hygiène et beauté » de l’autorité de régulation professionnelle de la publicité (ARPP), adoptée en octobre 2013. La DGCCRF peut s’appuyer sur ces règles d’autodiscipline pour introduire, si nécessaire, une action en publicité mensongère.
b. Les choix autorisés par le règlement
Si l’adaptation des normes nationales aux dispositions du règlement procède largement d’une simple retranscription, des marges de manœuvres ont été maintenues par le règlement : cette faculté de choix est utilisée en matière de réglementation de l’établissement des opérateurs économiques et en matière de mise en œuvre de la cosmétoviligilance.
L’alinéa 4 de l’article 3 maintient l’obligation de déclaration préalable à l’ANSM de l’ouverture d’un établissement de fabrication ou de conditionnement de produits cosmétiques, prévue par l’article L. 5131-2. Le considérant 56 du règlement prévoit en effet que les États membres conservent la possibilité de réglementer l’établissement d’opérateurs économiques dans le secteur des produits cosmétiques. L’alinéa 6 prévoit, au même article L. 5131-2, une obligation de communication de toute modification des éléments constitutifs de la déclaration.
Les modalités de présentation et de contenu de cette déclaration sont définies par décret en Conseil d’État, conformément au 1° de l’article L. 5131-8 à l’alinéa 19 du présent article.
En revanche, cette obligation est supprimée en cas d’importation de produits cosmétiques, car la notification à la commission européenne, préalable à leur mise sur le marché concerne également les produits importés au sein de l’Union européenne. Doivent notamment être notifiés le nom et l’adresse de la personne responsable, qui est l’importateur dans ce cas, ainsi que le pays d’origine du produit. Les autorités compétentes des États membres ayant un droit d’accès aux informations notifiées, la déclaration à l’ANSM des établissements d’importation paraît redondante avec la notification communautaire alors même que le point d’entrée réel dans l’Union est sans incidence sur la sécurité du produit.
L’alinéa 12 du présent article fait désormais figurer à l’article L. 5131-5 le système de cosmétovigilance qui était précédemment défini à l’article L. 5131-9, abrogé en conséquence.
Ce système national complète un système européen prévu à l’article 23 du règlement et fondé sur la double obligation :
– pour la personne responsable ou le distributeur, de notifier sans délai à l’autorité nationale compétente tous les effets indésirables graves dont elle a connaissance ;
– pour l’autorité nationale compétente, d’informer ses homologues européennes de tous les effets indésirables graves qui lui sont notifiés, tant par des personnes responsables ou des distributeurs que par des professionnels de santé, des utilisateurs ou des consommateurs.
Selon l’article 2 du règlement, un « effet indésirable » est une « réaction nocive pour la santé humaine imputable à l’utilisation normale ou raisonnablement prévisible d’un produit cosmétique ».
Un « effet indésirable grave » est un effet indésirable « entraînant une incapacité fonctionnelle temporaire ou permanente, un handicap, une hospitalisation, des anomalies congénitales, un risque vital immédiat ou un décès ».
Pour les personnes responsables et les distributeurs de produits cosmétiques, l’alinéa 12 du présent article définit désormais l’obligation de notification des effets indésirables graves par renvoi à l’article 23 du règlement européen, ce qui emporte obligation de notifier directement les effets indésirables graves à l’ANSM. Cette obligation de déclaration des effets indésirables graves était précédemment satisfaite en cas de signalement aux services de la DGCCRF, charge à ces derniers d’en informer l’ANSM (6e alinéa de l’article L. 5131-9).
En outre, le règlement n’établit de distinction qu’entre les effets indésirables graves et les autres effets indésirables. Pourtant, l’alinéa 12 prévoit qu’en complément des obligations découlant du règlement européen, la personne responsable et le distributeur peuvent déclarer les « effets indésirables qui, bien que n’ayant pas le caractère d’effets indésirables graves au sens du règlement leur paraissent revêtir un caractère de gravité justifiant une telle déclaration ». Pour votre rapporteur, le maintien de cette catégorie distincte d’effets indésirables, reprise des dispositions actuelles de l’article L. 5131-9, ne semble pas conforme au règlement et paraît en outre privée d’effets, puisque la déclaration dans ce cas reste facultative, comme pour tous les effets indésirables autres que graves.
Précisions que l’article 23 du règlement européen prévoit que la notification des effets indésirables est effectuée « sans délai », notion précisée par les lignes directrices définies par la commission européenne selon lesquelles « en ce qui concerne l’interprétation du délai (…), il faut compter 20 jours civils à partir de la date à laquelle tout employé de l’entreprise, quel que soit son rôle ou sa fonction, prend connaissance de l’événement indésirable grave. ».
Par ailleurs, le règlement européen a laissé aux État membres la faculté de réglementer, dans le respect de la législation communautaire, la notification des effets indésirables graves par les professionnels de la santé.
L’alinéa 13 du présent article traduit le choix de maintenir en conséquence une obligation pour les professionnels de santé de notifier les effets indésirables graves, obligation qui figure déjà dans la rédaction actuelle de l’article L. 5131-9 désormais abrogé.
Mais le règlement n’autorise pas les États membres à instaurer une obligation de déclarer les autres effets indésirables. L’alinéa 13 précise donc que la déclaration des autres effets indésirables est une simple faculté pour les professionnels de santé.
De même, à l’alinéa 14, les utilisateurs professionnels de produits cosmétiques, s’ils ne sont pas professionnels de santé, ne sont pas soumis à l’obligation de déclaration des effets indésirables graves mais peuvent procéder à la déclaration de tout effet indésirable, prévue également, à l’alinéa 15, pour les consommateurs.
Enfin, il convient de préciser que le règlement européen distingue nettement les effets indésirables et les effets occasionnés par des mésusages. Si l’effet indésirable peut conduire à une reformulation du produit cosmétique, le mésusage peut nécessiter un changement de conditionnement du produit ou une modification des précautions d’emploi précisées sur l’étiquetage, conformément à l’article 19 du règlement européen. La déclaration des effets consécutifs d’un mésusage doit donc être distincte de celle des effets indésirables.
Le 3° de l’article L. 5131-8 à l’alinéa 21 renvoie à un décret en Conseil d’État les modalités de mise en œuvre de ce système de cosmétovigilance.
En outre, l’alinéa 16 du présent article, supprime, à l’article L. 5131-6, les dispositions relatives aux informations, notamment d’étiquetage, devant figurer sur le produit, désormais directement définies par le règlement européen. Dans sa nouvelle rédaction, l’article L. 5131-6 définit désormais les mesures visant à renforcer les pouvoirs de contrainte de l’ANSM pour obtenir des informations quant à la composition des produits cosmétiques en cas de doute sur la sécurité d’une substance. L’ANSM pourra assortir d’une astreinte la demande motivée, rebaptisée « mise en demeure », afin d’obtenir de la personne responsable la liste des produits contenant la substance suspecte, ainsi que sa quantité pour chacun des produits concernés. L’astreinte peut atteindre 500 euros par jour de retard et à un montant maximal de 15 000 euros. Ce dispositif, gage d’efficacité, remplace le mécanisme actuel visant le même objectif, défini par l’article L. 5131-10, abrogé, qui ne comporte pas de pouvoir de sanction administrative.
Il revient également aux autorités nationales de préciser les modalités d’étiquetage : le 2° de l’article L. 5131-8 à l’alinéa 20 prévoit qu’un décret en Conseil d’État définit ces modalités après avis du Conseil national de la consommation.
Enfin, aux alinéas 17 et 18, les dispositions actuelles de l’article L. 5131-7 sont supprimées, qui subordonnent la mise sur le marché d’un produit cosmétique à la transmission d’un dossier d’information aux centres antipoison. À nouveau, la notification centralisée à la commission européenne justifie la mesure de simplification administrative. Dans sa nouvelle rédaction, l’article L. 5131-7 prévoit cependant la conservation de ces dossiers, par la personne responsable comme par le centre antipoison, jusqu’au 11 juillet 2020, pour tout produit cosmétique mis sur le marché avant la date d’entrée en vigueur du règlement européen. L’alinéa 95, au VI du présent article, prévoit, en conséquence, l’abrogation de cette disposition transitoire à compter du 12 juillet 2020.
c) Les sanctions pénales
L’article 37 du règlement européen prévoit que « les États membres déterminent le régime des sanctions applicables aux violations des dispositions du présent règlement et prennent toutes mesures nécessaires pour assurer la mise en œuvre de celles-ci. Les sanctions ainsi prévues doivent être effectives, proportionnées et dissuasives. »
Le II du présent article, aux alinéas 23 à 43 opère les adaptations rédactionnelles nécessaires, pour chacun des articles du code de la santé publique fixant les sanctions de la méconnaissance des règles régissant les produits cosmétiques. Le niveau des sanctions pénales n’est pas modifié mais les définitions des cas d’infractions sont adaptées aux nouvelles dispositions du règlement.
Les alinéas 24 à 28 modifient l’article L. 5431-2 : la peine de deux ans d’emprisonnement et de 30 000 euros d’amende est désormais encourue non seulement dans les cas d’ouverture d’établissement sans déclaration préalable à l’ANSM mais également en l’absence de notification à la commission européenne de l’ensemble des éléments exigés par l’article 13 du règlement, et, pour les nanomatériaux, par l’article 16, et enfin, en cas de violation des règles relatives aux expérimentations animales prévues par l’article 18 du règlement.
De même, c’est par renvoi aux définitions figurant dans le règlement, que les alinéas 29 à 31 adaptent la rédaction l’article L. 5431-5 actuel qui soumet à la même peine de deux ans d’emprisonnement et de 30 000 euros d’amende le fait de mettre sur le marché des produits cosmétiques non-conformes aux règles relatives aux substances entrant dans la composition du produit.
Les alinéas 32 à 35 opèrent des modifications du même ordre à l’article L. 5431-6 : la punition d’un an d’emprisonnement et de 15 000 euros d’amende est maintenue en cas d’infraction aux règles de tenue et de mise à disposition d’un dossier d’information sur le produit définies à l’article 11 du règlement. Les alinéas 37 à 39 modifient de la même façon l’article L. 5431-7 qui punit de 15 000 euros d’amende la méconnaissance des règles en matière d’étiquetage précisées à l’article 19 du règlement.
Les alinéas 41 et 42 ajoutent un article L. 5431-8, nouveau, qui sanctionne de deux ans d’emprisonnement et de 30 000 euros d’amende le fait pour la personne responsable et pour le distributeur de ne pas signaler les effets indésirables graves. La même peine est prévue dans le cas où un professionnel de santé méconnaît cette obligation.
Enfin l’alinéa 43 ajoute un article L. 5431-9, nouveau, en remplacement du 3° de l’article L. 5431-6, abrogé par l’alinéa 36 : il punit d’un an d’emprisonnement et de 15 000 euros d’amende le fait pour la personne responsable de ne pas transmettre à l’ANSM, en cas de doute sérieux sur la sécurité, les informations sur les substances qui composent le produit cosmétique. Le maintien de la sanction pénale s’ajoute donc à la sanction administrative de la mise en demeure sous astreinte, instituée par l’article L. 5131-6.
2. Les produits de tatouage
Les produits de tatouage sont définis à l’article L. 513-10-1 du code de la santé publique comme « toute substance ou préparation colorante destinée, par effraction cutanée, à créer une marque sur les parties superficielles du corps humain ».
Le premier alinéa de l’article L. 513-10-2 dans sa rédaction actuelle prévoit que sont applicables aux produits de tatouage les dispositions prévues pour les produits cosmétiques. Mais ces dernières sont modifiées par les mesures d’adaptation au règlement européen qui n’a pas pour objet de réglementer les produits de tatouage. Les modifications qu’il apporte ne sauraient donc être étendues, en tant que telles, aux produits de tatouage.
En conséquence, le III du présent article fait désormais figurer les dispositions existantes directement au chapitre X relatif aux produits de tatouage du titre III du livre Ier de la cinquième partie du code de la santé publique.
Le parti pris adopté consiste donc à maintenir des définitions et les procédures dans leur version actuelle. Les intervenants du système national de vigilance exercé sur les produits de tatouage sont en effet habitués à ces mécanismes et les coûts occasionnés par des modifications importantes de ces règles ne seraient pas compensés par des gains tirés de l’harmonisation au plan européen, qui n’existe pas encore.
L’article L. 513-10-2, aux alinéas 45 à 49, est remplacé par les dispositions figurant, pour les produits cosmétiques, à l’article L. 5131-2 dans sa rédaction actuelle : il revient à la personne responsable de la mise sur le marché d’adresser à l’ANSM des déclarations d’ouverture et d’exploitation des établissements fabriquant, conditionnant ou important des produits de tatouage. Les modalités de déclaration sont définies par décret, conformément au 1° de l’article L. 513-10-10, nouveau, à l’alinéa 69. La personne responsable doit également désigner des personnes qualifiées chargées de l’évaluation de la sécurité pour la santé humaine, dont les diplômes sont précisés par arrêté.
À l’alinéa 51, l’article L. 513-10-3 est modifié afin de clarifier les compétences de l’ANSM en matière de fixation des normes opposables aux fabricants.
D’une part, les dispositions prévoyant que les bonnes pratiques de fabrication des produits de tatouage restent définies par un arrêté interministériel, sur proposition de l’ANSM. Il en va de même pour les règles de délivrance des documents attestant le respect de ces bonnes pratiques.
Mais, concernant la définition des règles des bonnes pratiques de laboratoires, il y a suppression du renvoi à un arrêté interministériel pris sur proposition de l’ANSM, à l’exemple, en dernier lieu, d’un arrêté du 23 juin 2011 : l’agence les publie désormais directement en application des principes européens définis dans la directive 2004/10/CE relative aux bonnes pratiques des laboratoires, précédemment mentionnée. Il en va de même pour les règles d’inspection et de vérification du respect de ces bonnes pratiques.
L’alinéa 53 fait figurer à l’article L. 513-10-4, modifié, la définition de l’obligation, pour les produits de tatouage, de ne pas nuire à la santé humaine lorsqu’ils sont appliqués dans les conditions normales d’utilisation : elle reprend la rédaction de l’article L. 5131-4 dans sa rédaction actuelle. Le 6° de l’article L. 513-10-10, nouveau, à l’alinéa 74, prévoit que les règles en matière de qualité, de sécurité et de composition des produits sont déterminées par décret.
Les alinéas 55 à 57 introduisent un article L. 513-10-5, nouveau, qui reprend les dispositions actuellement applicables par renvoi à l’article L. 5131-6 : respect des obligations en matière d’étiquetage et mise à disposition d’un dossier d’information permettant de vérifier la composition du produit et l’évaluation de sa sécurité pour la santé humaine. Les 2° et 3° de l’article L. 513-10-10, nouveau, à aux alinéas 70 et 71, prévoient que des décrets déterminent respectivement les mentions que doivent comporter le récipient et l’emballage du produit de tatouage, et le contenu et les modalités de conservation du dossier d’information.
Les alinéas 58 à 59 maintiennent, à l’article L. 513-10-6, nouveau, la transmission aux centres antipoison d’ « informations adéquates et suffisantes concernant les substances utilisées », conformément à la rédaction actuelle de l’article L. 5131-7. À défaut d’une notification centralisée de ces informations au plan européen, le maintien de la compétence de cette instance de contrôle de proximité semble en effet nécessaire. Et l’alinéa 60 prévoit à l’article L. 513-10-7, nouveau, que la personne responsable met en outre à la disposition du public les informations liées à la composition et aux effets indésirables du produit, définies par décret, selon le 4° de l’article L. 513-10-10, à l’alinéa 72.
L’article L. 513-10-8, nouveau, établi par les alinéas 61 à 65, décrit le système national de vigilance exercé sur les produits de tatouage. Au regard du dispositif présenté pour les produits cosmétiques, les obligations de déclaration des effets indésirables sont systématisées, sans doute en raison du fait que les produits de tatouage font l’objet d’une injection et non d’une simple application, comme les produits cosmétiques.
Ainsi la personne responsable doit déclarer sans délai les effets indésirables graves ainsi que les autres effets indésirables. La définition spécifique des effets indésirables englobe les cas de mésusages. La déclaration peut être faite soit directement à l’ANSM, doit à la DGCCRF qui en informe alors l’agence nationale. Les professionnels de santé sont également soumis à l’obligation de déclarer l’ensemble des effets indésirables dont ils ont connaissance. Cette obligation est en outre étendue à toute personne qui réalise des tatouages à titre professionnel. Enfin, les consommateurs ont la faculté de déclarer les effets indésirables auxquels ils sont sujets. Le 5° de l’article L. 513-10-10 nouveau, à l’alinéa 73, prévoit que les modalités de mise en œuvre de ce système national de vigilance sont précisées par décret.
Enfin, les alinéas 66 et 67 établissent un article L. 513-10-9, nouveau, qui maintient la compétence de l’ANSM pour adresser au producteur ou à l’importateur une demande motivée d’informations sur les substances utilisées en cas de doutes sérieux sur leur innocuité.
• Les sanctions pénales
Les infractions et peines relatives aux produits de tatouage sont actuellement définies à l’article L. 5437-2 par renvoi aux dispositions des articles L. 5431-2, L. 5431-3 et L. 5431-4 relatifs aux cosmétiques, dans leur rédaction actuelle. Le IV du présent article établit directement ces dispositions dans un chapitre VII relatif aux produits de tatouage, du titre III du livre IV, relatif aux sanctions pénales et financières, de la cinquième partie du code de la santé publique.
Les alinéas 76 à 80 modifient l’article L. 5437-2 pour y transcrire les dispositions de l’article L. 5431-2, maintenues : est ainsi passible d’une peine de deux ans d’emprisonnement et de 30 000 euros d’amende, la méconnaissance des obligations de déclaration d’un établissement produisant ou important des produits de tatouage, de désignation d’une personne qualifiée responsable de la sécurité des produits et de transmission des informations adaptées aux centres antipoison.
Les alinéas 82 à 87 transposent à l’article L. 5437-3, nouveau, les peines complémentaires prévues à l’article L. 5431-3 applicables aux personnes physiques : diffusion de la condamnation, affichage de la décision, confiscation du produit de la vente de la chose destinée à commettre l’infraction, fermeture des établissements ayant servi à commettre les faits incriminés. Les alinéas 88 à 91 transcrivent les peines applicables aux personnes morales, fixées par l’article L. 5431-4, dans un nouvel article L. 5437-4.
Enfin, l’article L. 5437-5, nouveau, à l’alinéa 92, sanctionne de deux ans d’emprisonnement et de 30 000 euros d’amende, le fait pour la personne responsable de la mise sur le marché de ne pas déclarer à l’ANSM les effets indésirables graves du produit de tatouage dont elle a connaissance. L’alinéa 93 prévoit la même sanction en cas de méconnaissance de cette obligation par les professionnels de santé ou les personnes qui réalisent des tatouages à titre professionnel.
*
La Commission examine d’abord l’amendement AS9 du rapporteur.
M. le rapporteur. L’article 10 du règlement européen prévoit que l’évaluation de la sécurité est effectuée par une personne titulaire soit d’un diplôme ou d’un autre titre sanctionnant une formation universitaire d’enseignement théorique et pratique en pharmacie, toxicologie, médecine ou dans une discipline analogue, soit d’une formation reconnue équivalente par un État membre.
L’article 3 précise qu’un arrêté définit les formations reconnues équivalentes en France, mais celui-ci ne saurait faire obstacle à la reconnaissance automatique des formations définies comme équivalentes dans d’autres États membres. Il convient donc que la loi soit plus précise à cet égard.
Reste qu’un État membre qui reconnaîtrait comme équivalente aux diplômes mentionnés par le règlement européen des formations manifestement insuffisantes se trouverait en infraction au regard du droit communautaire.
La Commission adopte l’amendement.
Elle examine ensuite l’amendement AS5 du rapporteur.
M. le rapporteur. L’article 3 prévoit qu’en complément des obligations découlant du règlement européen, la personne responsable du produit cosmétique et son distributeur peuvent déclarer les « effets indésirables qui, bien que n’ayant pas le caractère d’effets indésirables graves au sens du règlement, leur paraissent revêtir un caractère de gravité justifiant une telle déclaration ».
Je vous propose de supprimer cette notion, car le règlement européen n’établit de distinction qu’entre les effets indésirables graves et les autres effets indésirables et que cette catégorie intermédiaire, non prévue par le règlement, serait dénuée de portée.
La Commission adopte l’amendement.
Elle examine ensuite, en discussion commune, les amendements AS6, AS7 et AS8 du rapporteur.
M. le rapporteur. Le règlement européen définit l’effet indésirable comme « une réaction nocive pour la santé humaine imputable à l’utilisation normale ou raisonnablement prévisible d’un produit cosmétique ».
Les cas de mésusage sont donc distincts des effets indésirables, qui sont les seuls à relever des prescriptions du règlement.
Je vous propose donc de mieux distinguer la déclaration des effets susceptibles de résulter d’un mésusage.
La Commission adopte les amendements AS6, AS7 et AS8.
Elle adopte ensuite les amendements AS13, AS27, AS31, AS14 et AS15 rédactionnels du rapporteur.
Elle en vient à l’amendement AS3 du rapporteur.
M. le rapporteur. Cet amendement vise à préciser le délai dans lequel les fabricants et distributeurs de produits cosmétiques doivent déclarer les effets indésirables graves à l’ANSM.
L’article 3 prévoit actuellement que la déclaration a lieu « dès qu’ils en ont connaissance ». Il est proposé d’indiquer que la déclaration est effectuée « sans délai », notion précisée par la commission européenne dans ses lignes directrices : 20 jours civils à partir de la date à laquelle tout employé de l’entreprise, quel que soit son rôle ou sa fonction, prend connaissance de l’événement indésirable. Cela permet d’éviter une contestation du point de départ du délai.
La Commission adopte l’amendement.
Puis elle adopte les amendements AS16, AS17, AS18, AS19, AS20, AS21, AS22 et AS30 rédactionnels du rapporteur.
Elle examine ensuite l’amendement AS4 du rapporteur.
M. le rapporteur. Les professionnels de santé déclarent aujourd’hui les effets indésirables des tatouages qu’ils constatent, sans précisions sur les conditions de réalisation de ces tatouages.
J’ai auditionné les représentants du syndicat des tatoueurs, qui m’ont fait part d’un problème qui les mobilise depuis plusieurs jours : un projet d’arrêté par l’ANSM – dont l’application est prévue pour le 1er janvier prochain – tendant à interdire l’utilisation d’un certain nombre de pigments par technique d’effraction cutanée, correspondant à celle du tatouage. Ces pigments permettent de produire des colorants que l’on trouve dans des produits cosmétiques d’application cutanée mais qui sont utilisés régulièrement en Europe par les fabricants de produits de tatouage. Or une alerte a été lancée en 2008 par des dermatologues au niveau européen, qui a été reprise par le Conseil de l’Europe – ce qui a conduit un certain nombre de pays à modifier les autorisations en vue de fabriquer des colorants de produits de tatouage. Les tatoueurs s’alarment du fait que l’arrêté diminuera le nombre de coloris disponibles et souhaitent attendre une décision de l’agence européenne de sécurité sanitaire, prévue dans quelques mois. J’ai interpellé la direction générale de la santé et l’ANSM à cet égard.
Par ailleurs, j’ai proposé aux tatoueurs que, en cas de déclaration d’effet indésirable grave survenu après un tatouage, il y ait une enquête systématique portant sur la nature du produit utilisé – pour vérifier qu’il ne soit pas contrefait – ainsi que sur les compétences du tatoueur et les conditions dans lesquelles le tatouage a été réalisé – pour vérifier que celui-ci a été effectué par un tatoueur agréé. Tel est le sens du présent amendement.
La Commission adopte l’amendement.
Elle adopte ensuite les amendements AS23, AS28, AS24, et AS25 rédactionnels du rapporteur.
Puis elle adopte l’article 3 modifié.
Article 4
(art. L. 4 362-9-1 [nouveau] et L. 4363-4 du code de la santé publique)
Vente sur internet de lentilles correctrices
Le présent article vise à reconnaître la commercialisation en ligne de lentilles correctrices d’amétropie et à mettre ainsi un terme à une procédure d’infraction à la législation européenne entamée par la commission européenne à l’encontre de la France le 27 juin 2007.
Comme les verres de lunettes, les lentilles correctrices sont des dispositifs médicaux destinés à corriger les « amétropies », c’est-à-dire les défauts de vision. Les lentilles correctrices sont cependant des dispositifs médicaux invasifs, relevant de la classe de risques IIA définie par la directive 93/42 CEE du 14 juin 1993 regroupant les dispositifs médicaux présentant un degré moyen de risque.
Le présent article vise donc également à encadrer la vente en ligne de lentilles afin de protéger la santé des utilisateurs.
1. Une vente en ligne autorisée par le droit européen
La vente en ligne de lentilles correctrices intéresse à la fois la libre circulation des biens et services et la réglementation du commerce électronique.
Le principe de libre-circulation des marchandises et des services est consacré par le traité sur le fonctionnement de l’Union européenne (TFUE). La libre-circulation des marchandises repose essentiellement sur l’interdiction, à l’article 34, des restrictions quantitatives à l’importation et des mesures d’effets équivalents. Il est toutefois possible de recourir à de telles restrictions pour un certain nombre de motifs énumérés par l’article 36 du TFUE, notamment afin d’assurer la protection de la santé et de la vie des personnes.
Le commerce électronique est régi par le droit dérivé de l’Union européenne. Le rapprochement des législations des États-membres en ce domaine est organisé par la directive n° 2000/31/CE du 8 juin 2000, relative à certains aspects juridiques des services de la société de l’information, et notamment du commerce électronique. Les États-membres doivent s’assurer que l’accès à une activité de service de la société d’information n’est soumis à aucune autorisation préalable et que leur système juridique rendent possible la conclusion de contrats par voie électronique.
Les seules restrictions possibles à cette libre-circulation concernent les mesures nécessaires et proportionnées à un objectif d’ordre public, de protection de la santé publique, de sécurité publique ou de protection des consommateurs.
La Cour de justice de l’Union européenne a eu à connaître d’un règlement hongrois interdisant la vente en ligne de lentilles de contact. Dans l’arrêt KER-Optika du 2 décembre 2010, la Cour a considéré que les articles 34 et 36 du TFUE ainsi que la directive 2000/31 s’opposent à une réglementation nationale qui n’autorise la commercialisation de lentilles de contact que dans des magasins spécialisés en dispositifs médicaux.
L’interdiction de livraison de lentilles commandées en ligne est en effet une mesure d’effet équivalent à une restriction quantitative d’importation ; et cette mesure ne saurait être justifiée par l’objectif de protection de la santé car ce dernier peut être atteint par d’autres moyens moins restrictifs que l’interdiction de vente en ligne.
La Cour a donc précisé qu’ « un État membre peut exiger que les lentilles de contact soient délivrées par un personnel qualifié qui attire l’attention du client sur ces risques, procède à des examens du client et recommande ou déconseille à ce dernier le port de lentilles, en invitant l’intéressé, le cas échéant, à consulter un médecin ophtalmologiste. »
Or ces mesures peuvent être mises en œuvre dans le cadre de la vente par internet. De fait, les conseils peuvent être fournis à distance par des moyens interactifs et l’adaptation des lentilles, qui peut être réalisée par un ophtalmologiste, n’est nécessaire que lors de la première délivrance de lentilles.
Il en résulte que l’interdiction de livraison de lentilles commandées par internet est contraire à la libre circulation des marchandises dans l’Union.
La Cour a cependant précisé que les règles nationales relatives aux modalités de délivrance des lentilles correctrices elles-mêmes, qu’elles soient achetées sur internet ou en boutique, n’entrent pas dans le champ de la directive : il revient donc aux seules autorités compétentes des États membres de déterminer les conditions de délivrance de ces dispositifs médicaux.
2. La nécessité d’adapter la législation française
La vente en ligne de lentilles correctrices n’est actuellement ni reconnue ni interdite par la législation française. Aujourd’hui de nombreux sites de vente en ligne permettent pourtant d’acheter des lentilles correctrices.
Le décalage entre le droit et la pratique est une source d’insécurité juridique qui fragilise l’exercice d’un droit reconnu par les traités européens et la jurisprudence de la Cour.
La Commission européenne a, en conséquence, engagé une procédure d’infraction à l’encontre de la France pour « entraves à la commercialisation des lentilles de contact » (procédure n° 2005/5070). Après une mise en garde le 27 juin 2007, un avis motivé a été adressé à la République française, le 18 septembre 2008, afin de l’inviter à préciser la législation en vigueur.
La commission a ainsi relevé qu’en l’absence de précision, l’interdiction du « colportage des verres correcteurs d’amétropie », figurant à l’article L. 4362-9 du code de la santé publique peut être interprétée comme n’autorisant la vente de ces dispositifs médicaux que dans des magasins d’optique ce qui, d’après la jurisprudence KER-Optika, serait contraire à la libre-circulation des marchandises et au libre accès aux services de la société de l’information.
La procédure d’infraction, première étape du recours en manquement, peut donner lieu à tout moment à une saisine de la Cour de justice par la Commission européenne.
La menace d’une condamnation de la France est donc réelle. Si le constat d’un développement important de la vente en ligne de lentilles correctrices depuis 2007 atténue la portée des observations de la commission, l’absence de saisine de la Cour de justice tient principalement au fait qu’une mise en conformité a été initiée en 2011, qui n’a pas été menée à terme.
Lors de l’examen du projet de loi renforçant les droits, la protection et l’information du consommateur (n° 3508), déposé sur le bureau de l’Assemblée nationale le 1er juin 2011, il a été envisagé d’insérer dans le code de la santé publique un nouvel article L. 4 362-9-1 qui reconnaît la vente de lentille en ligne et impose aux prestataires concernés de mettre à disposition des patients un opticien-lunetier. L’article L. 4363-4 du même code était modifié afin de sanctionner les infractions à cette obligation d’une amende de 3 750 euros.
Cependant, l’examen de ce projet de loi n’a pas été poursuivi au-delà de la première lecture.
Une nouvelle occasion de mise en conformité de notre droit s’est présentée en 2013, lors de l’examen du projet de loi relatif à la consommation (n° 1015), déposé sur le bureau de l’Assemblée nationale le 2 mai 2013.
Lors de la première lecture au Sénat en septembre 2013, un amendement du rapporteur a introduit un nouvel article 17 quater reprenant l’essentiel du dispositif proposé en 2011 : cet article insère un nouveau chapitre V « Verres correcteurs et lentilles de contact oculaire correctrices » dans le titre Ier du livre II de la partie V du code de la santé publique, constitué d’un article unique (article L. 5215-1 nouveau), qui traite spécifiquement de la vente en ligne de lentilles de contact oculaire correctrices mais également de verres correcteurs. Cet article régit les conditions de vente en cas de recours « à une technique de communication à distance ». Il prévoit que le vendeur doit mettre à disposition du patient un « opticien-lunetier ». Le non-respect de cette obligation serait sanctionné par une amende de 10 000 euros (article L. 5461-6-1 nouveau).
Cet amendement a été adopté au Sénat postérieurement au dépôt, sur le bureau de l’Assemblée nationale, le 2 août 2013, du présent projet de loi, dont l’article 4 vise le même objectif mais pour les seules lentilles correctrices et selon un dispositif légèrement différent.
3. Une légalisation doublée de mesures d’encadrement
• Le dispositif projeté
Le I insère dans le code de la santé publique, un article L. 4 362-9-1 nouveau qui reconnaît et encadre la vente en ligne de lentilles correctrices.
L’alinéa 3, au I de l’article L. 4 362-9-1 prévoit que « lors de la vente en ligne de lentilles correctrices, les prestataires concernés permettent au patient d’obtenir informations et conseils auprès d’un professionnel de santé qualifié ».
Le terme de « lentilles correctrices » recouvre à la fois les lentilles dites « de contact », qui reposent sur la cornée, et également les « verres scléraux », lentilles qui reposant sur la conjonctive, membrane transparente qui recouvre la sclère, c’est-à-dire le « blanc » de l’œil.
Il s’agit d’exiger, pour la vente en ligne, la mise à disposition du client d’informations et de conseils émanant d’un « professionnel de santé qualifié ». La définition des mentions et informations devant figurer sur le site internet sont renvoyées à un décret en Conseil d’État.
Votre rapporteur estime en effet indispensable que l’acheteur en ligne dispose de toutes les informations nécessaires. À ce titre, la vente en ligne peut représenter un avantage, si le site du vendeur permet à l’utilisateur de formuler à distance des questions, même très détaillées, auxquelles des professionnels responsables du service de support du site apporteraient les réponses adaptées.
La mention d’un « professionnel de santé qualifié » est plus large que celles des seuls opticiens-lunetiers : la dispensation de conseils et informations aux patients peut en effet être assurée, par un ophtalmologiste et selon le champ de compétence, par un orthoptiste ou un opticien-lunetier.
L’alinéa 2, au I de l’article L. 4 362-9-1, prévoit que « les conditions de première délivrance de lentilles correctrices sont déterminées par décret en Conseil d’État ».
Votre rapporteur estime qu’une ordonnance médicale adaptée au porteur semble en effet nécessaire à la première délivrance car seule une consultation médicale avec examen physique des yeux est en mesure d’écarter les contre-indications au port de lentilles et de déterminer les différents paramètres des lentilles (matériau, modèle, diamètre, rayon de courbure, puissance).
Selon l’étude d’impact jointe au projet de loi, le décret imposerait de s’assurer, à la première délivrance de lentilles correctrices, de la réalisation préalable de l’adaptation des lentilles et de la validité de l’ordonnance pour les mineurs de moins de seize ans.
L’alinéa 4, au II du présent article, modifie l’article L. 4363-4 qui sanctionne d’une amende de 3 750 euros le non-respect des règles relatives à l’optique-lunetterie. Un 4° est ajouté afin de sanctionner le non-respect des conditions de première délivrance des lentilles correctrices ou l’absence de mise à disposition de conseils émanant de professionnels lors de la vente en ligne. Un tel dispositif de sanction dispense d’instaurer une procédure d’autorisation préalable du site internet ou de déclaration aux autorités sanitaires.
Les conditions de commercialisation des lentilles correctrices proposées par le présent article permettent donc de concilier la reconnaissance de la légalité de la vente en ligne et la protection de la santé des patients.
• Les effets attendus de diminution du reste à charge pour les assurés
Votre rapporteur souhaite souligner que l’encadrement de la vente en ligne de lentilles correctrices, en sécurisant l’accès à ces produits, va contribuer à améliorer l’offre en ligne, ce qui devrait entraîner une baisse des prix.
Dans son rapport de septembre 2013 sur l’application des lois de financement de la sécurité sociale, la Cour des comptes a examiné les modalités de prise en charge par les organismes de protection sociale de l’optique correctrice. La Cour a constaté que les dépenses d’optique connaissent une croissance soutenue et que les prix de l’optique correctrice demeurent élevés alors même qu’ils devraient diminuer sous l’effet de la baisse des coûts de production. Les dépenses d’optique par habitant seraient deux fois supérieures en France par rapport au reste de l’Union européenne : 88 euros par habitant et par an, contre 54 euros en Allemagne, 49 euros au Royaume-Uni, 36 euros en Italie et 30 euros en Espagne.
Cette situation résulterait selon la Cour de l’opacité du marché, trop peu concurrentiel, et de la faible diffusion en France des nouveaux modes de distribution. Selon la Cour, « le développement du commerce en ligne pour le choix de la monture, la commande des verres et des lentilles correctrices [pourrait] utilement contribuer au renforcement de la concurrence ». Ces modes de distribution auraient permis de réduire le coût des produits d’optique au Royaume-Uni ou en Allemagne.
Votre rapporteur se félicite donc que la mise en conformité avec le droit européen contribue, à terme, à une diminution du reste à charge pour les assurés.
*
La Commission adopte l’article sans modification.
Article 5
(ordonnance n° 2012-1427 du 19 décembre 2012, art. L. 5124-1, L. 5125-33, L. 5125-34, L. 5125-39, L. 5438-2, L. 5438-6, L. 5438-7, L. 5438-8 [nouveau] du code de la santé publique)
Lutte contre la falsification des médicaments et encadrement de la vente en ligne par des pharmaciens d’officine
Cet article vise à transposer la directive 2011/62/UE du Parlement européen et du Conseil du 8 juin 2011 qui a modifié la directive 2001/83/CE du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain en ce qui concerne la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés.
Cette directive, qui porte notamment sur la vente de médicaments à distance au public, a été transposée en droit français par une ordonnance n°2012-1427 du 19 décembre 2012, prise sur le fondement de l’article 38 de la Constitution, ainsi que par un décret n° 2012-1562 du 31 décembre 2012.
Le I du présent article ratifie l’ordonnance n° 2012-1427 du 19 décembre 2012 relative au renforcement de la sécurité de la chaîne d’approvisionnement des médicaments, à l’encadrement de la vente de médicaments sur internet et à la lutte contre la falsification des médicaments. Le II y apporte quelques modifications.
1. La ratification de l’ordonnance n° 2012-1427 du 19 décembre 2012
L’article 38 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé a habilité le Gouvernement à prendre par ordonnance les mesures nécessaires à la transposition de la directive dite « Médicaments falsifiés ». Cette directive a introduit des mesures de sécurité et de contrôle harmonisées à l’échelle de l’Europe visant à repérer plus facilement les médicaments falsifiés et à procéder à des vérifications et contrôles plus rigoureux sur le territoire des États-membres comme aux frontières de l’Union.
La directive donne tout d’abord une définition précise du médicament falsifié, entendu comme « tout médicament comportant une fausse présentation de :
a) son identité, y compris de son emballage et de son étiquetage, de sa dénomination ou de sa composition s’agissant de n’importe lequel de ses composants, y compris les excipients, et du dosage de ces composants ;
b) sa source, y compris de son fabricant, de son pays de fabrication, de son pays d’origine ou du titulaire de son autorisation de mise sur le marché ;
c) son historique, y compris des enregistrements et des documents relatifs aux circuits de distribution utilisés. »
Cette définition « n’inclut pas les défauts de qualité non intentionnels et s’entend sans préjudice des violations des droits de propriété intellectuelle ».
La directive met également en place un régime juridique précis pour les excipients, qui sont désormais définis comme « tout composant d’un médicament, autre qu’une substance active et les matériaux d’emballage », et à l’égard desquels des lignes directrices sur les bonnes pratiques de fabrication sont adoptées. Possibilité est laissée aux États membres d’étendre les dispositifs de sécurité assurant l’inviolabilité des emballages à tous les médicaments. Ceci permettra, en France, de mettre en place un film protecteur sur l’emballage des médicaments.
En outre, le Gouvernement a été habilité à prendre les mesures visant à encadrer l’information et le commerce électroniques concernant l’ensemble des produits à finalité sanitaire.
L’ordonnance n° 2012-1427 a été publiée le 19 décembre 2012. Conformément à son article 23, ses dispositions sont entrées en vigueur le 1er avril 2013. La loi d’habilitation prévoyait le dépôt devant le Parlement du projet de loi de ratification de l’ordonnance dans un délai de quatre mois suivant la publication de cette dernière, ce qui correspondait au 19 avril 2013. Le présent projet de loi ayant été déposé le 2 août 2013, on peut regretter un retard, mais il reste bien moindre que celui que l’on constate habituellement. Ce délai supplémentaire est au demeurant pleinement justifié par l’intervention, le 14 février 2013, du juge des référés du Conseil d’État (cf. infra).
2. Les dispositions de l’ordonnance n° 2012-1427 du 19 décembre 2012
Le I du présent article ratifie l’ordonnance dont les différents articles visent à transposer l’ensemble des objectifs de la directive.
• La définition des médicaments falsifiés
L’article 2 de l’ordonnance ajoute ainsi un article L. 5111-3 nouveau au code de la santé publique qui définit le médicament falsifié comme tout médicament « comportant une fausse présentation » qui peut porter sur des éléments :
– relatifs à l’identité du produit, ce qui comprend l’ensemble des éléments entrant dans sa composition ;
– explicitant sa source : son fabriquant, son pays de fabrication, etc. ;
– et enfin portant sur son historique, cette notion incluant les autorisations, enregistrements et les documents relatifs aux circuits de distribution utilisés.
Cette définition de la fausse présentation paraît complète et exclut les défauts de qualité non intentionnels.
• La réglementation des activités de courtage en médicament
L’article 6 de l’ordonnance définit à l’article L. 5124-19 l’activité de courtage comme « toute activité liée à la vente ou à l’achat de médicaments qui ne comprend pas de manipulation physique et qui consiste à négocier, indépendamment ou au nom d’une personne physique ou morale ». Cette activité doit être déclarée auprès de l’agence nationale de sécurité du médicament et des produits de santé (ANSM) dès lors que le courtier exerce en France.
Les courtiers ne peuvent exercer leur activité que sur des médicaments ayant obtenu une autorisation de mise sur le marché ou encore un « enregistrement au titre des articles L. 5121-13 ou L. 5221-14-1 », ce qui correspond au régime de commercialisation de la plupart des médicaments homéopathiques ou de médicaments naturels à base de plantes.
Aussi, le 1° de l’article 14 inclut les activités de courtage dans le champ de compétence de l’ANSM. Auditionné par votre rapporteur, le directeur général de l’ANSM a souligné l’intérêt de la réglementation des activités de ces intervenants exclusivement financiers de la chaîne du médicament. Leurs activités ne se traduisent pas par des mouvements physiques des lots de médicaments, mais le contrôle de leurs comptes renforce la traçabilité des produits et permet de repérer les montages frauduleux liés aux activités des faussaires.
À des fins de coordination, l’article 5 de l’ordonnance ajoute un alinéa à l’article L. 5124-1 au code de la santé publique afin d’exclure les courtiers en médicaments du champ d’application du chapitre portant sur la fabrication et la distribution en gros de produits pharmaceutiques. L’article 1er de l’ordonnance insère un article L. 4211-1-1 qui exclut ces mêmes personnes du champ d’application du titre premier du livre Ier de la quatrième partie du code de la santé qui définit l’exercice des professions médicales. Le 1°de l’article 3 modifie l’article L. 5121-5 qui porte sur les médicaments génériques : il élargit son champ aux activités de courtage en médicaments et précise explicitement qu’il recouvre les cas où le commerce de ces produits est assuré par voie électronique.
Par ailleurs, l’article 4 précise à l’article L. 5121-20 que l’activité de conditionnement est encadrée par un décret pris en Conseil d’État, au même titre que sont ainsi déterminés l’étiquetage, la notice ou encore la dénomination des médicaments.
• Le commerce électronique de médicaments
À l’article 7, le II insère un chapitre V bis au titre II du livre Ier de la cinquième partie du même code intitulé « Commerce électronique de médicaments par une pharmacie d’officine ». L’article L. 5125-33 définit l’activité de commerce électronique de médicaments. Elle doit répondre aux critères suivants :
– elle propose au public des médicaments à usage humain, accompagnés d’informations de santé ;
– elle n’est possible qu’à partir du site internet d’une officine de pharmacie, l’article L. 5125-35 précisant au demeurant que la création du site est subordonnée à l’existence de la licence de l’officine.
La création et l’exploitation du site sont réservées aux titulaires d’une officine ou gérant d’une pharmacie mutualiste ou de secours minière.
Le site internet de la pharmacie est donc considéré comme le prolongement virtuel d’une officine de pharmacie autorisée et ouverte au public. L’article L. 5125-37 prévoit ainsi qu’en cas de regroupement de plusieurs officines en un lieu unique, il ne peut être exploité qu’un seul site internet. Et l’article L. 5425-38 précise que la cessation d’activité de l’officine entraîne la fermeture du site.
L’article L. 5125-41 renvoie à un décret en Conseil d’État la définition des informations minimales contenues par le site internet. Par ailleurs, le 2°de l’article 3 de l’ordonnance précise au troisième alinéa de l’article L. 5121-5 que la dispensation de médicaments « y compris par voie électronique » doit être réalisée en conformité avec des bonnes pratiques dont les principes sont définis par arrêté du ministre chargé de la santé.
L’article 7 de l’ordonnance prévoit que l’article L. 5425-36 donne au directeur général de l’agence régionale de santé (ARS) de ressort compétence pour autoriser la création du site internet dédié. L’article L. 5125-39 prévoit en outre les conditions dans lesquels le directeur général de l’ARS peut intervenir en cas de manquement aux règles applicables au commerce électronique de produits pharmaceutiques. (8)
Aux termes de l’article L. 5125-34, établi par l’article 7 de l’ordonnance, n’étaient éligibles au commerce électronique que les seuls médicaments pouvant être présentés en accès direct au public en officine, après obtention de l’autorisation de mise sur le marché ou enregistrement. Il s’agit des médicaments figurant sur la liste, établie par l’ANSM, des médicaments dits de « médication officinale », à des fins d’automédication. Cette disposition a été abrogée par la décision n° 365317 du 17 juillet 2013 du Conseil d’État (cf. infra).
L’article L. 5125-40 autorise la vente en ligne par « une personne physique ou morale légalement habilitée à vendre des médicaments au public dans l’État membre de l’Union européenne dans laquelle elle est installée ». L’opérateur n’est donc pas forcément, dans ce cas, soumis à la même réglementation que les pharmaciens d’officine opérant depuis la France. Mais conformément au c) du paragraphe 1 à l’article 85 quater de la directive 2011/83/CE, l’opérateur d’un État membre qui vend des médicaments par internet à destination d’un autre État membre ne peut vendre que les médicaments autorisés à être vendus sur internet dans cet État. L’article L. 5125-40 restreint donc la vente aux catégories de médicaments définies à l’article L. 5125-34.
Il prévoit en outre le « respect de la législation applicable aux médicaments commercialisés en France » : cette mention paraît plus large que la lettre de la directive et pourrait être interprétée comme allant au-delà du fait, pour le médicament, d’avoir bénéficié d’une autorisation de mise sur le marché en France (ou de l’enregistrement applicable aux médicaments homéopathiques ou traditionnels à base de plantes). Cette mention pourrait donc utilement être revue afin de ne pas susciter de difficultés d’interprétations.
Le I de l’article 7 ajoute un article L. 5122-6-1 qui élargit au commerce de produits pharmaceutiques le champ d’application des dispositions du chapitre relatif à la publicité des produits, c’est-à-dire, au sens de l’article L. 5122-1, « toute forme d’information, y compris le démarchage, de prospection ou d’incitation qui vise à promouvoir la prescription, la délivrance, la vente ou la consommation de ces médicaments ».
En complément de l’article 7, l’article 18 de l’ordonnance modifie l’article L. 4211-1 du code de la santé publique afin d’élargir à la vente en ligne l’exclusivité de la vente de produits pharmaceutiques dévolue aux pharmaciens.
Enfin, le II de l’article 7 établit un article L. 5125-39 qui détermine le régime de sanction applicable en cas de manquement aux règles relatives au commerce électronique de médicaments. L’amende administrative qui peut être prononcée à l’encontre de l’auteur du manquement ne peut excéder 30 % du chiffre d’affaires réalisé par la pharmacie dans le cadre de l’activité de commerce électronique, dans la limite d’un million d’euros. Dans sa décision n° 365317 du 17 juillet 2013 précitée, le Conseil d’État a jugé que ce régime est conforme aux principes de légalité et de proportionnalité des peines. Le Conseil d’État a relevé que le seuil de 30 % du chiffre d’affaires annuel ne peut être considéré comme manifestement disproportionné au regard des manquements que la sanction a pour objet de réprimer. En outre, cette sanction fait l’objet d’une appréciation par le directeur général de l’ARS territorialement compétente, après que l’intéressé a été mis en mesure de présenter ses observations.
• La lutte contre la falsification des matières premières
L’article 9 de l’ordonnance établit au I de l’article L. 5138-2 du code de la santé publique une nouvelle rédaction qui précise la définition des matières premières à usage pharmaceutique. Elle distingue d’une part les substances actives, c’est-à-dire toute substance utilisée pour la fabrication d’un médicament et qui devient un composant actif de ce médicament, et d’autre part les excipients, c’est-à-dire les composants d’un médicament autre qu’une substance active et que les matériaux d’emballage.
L’article 13 insère un article L. 5138-6 au même code afin d’étendre la définition de la falsification des médicaments figurant à l’article L. 5111-3 aux matières premières des médicaments.
L’article 8 précise à l’article L. 5138-1 que la fabrication, l’importation et la distribution des substances actives comme des excipients ne peuvent être exercées que dans des établissements autorisés par l’ANSM et qui lui déclarent leur activité. Ceci inclut les activités menées en vue de l’exportation.
L’article 10 modifie l’article L. 5138-3. La fabrication des substances actives est soumise aux bonnes pratiques dont les principes sont définis par le directeur général de l’ANSM. Les établissements fabricant des médicaments doivent notamment veiller à la qualité et à l’authenticité des matières premières employées, n’utiliser que des matières premières fabriquées et distribuées selon les bonnes pratiques, ou encore se conformer à des obligations d’audits.
Un article L. 5138-3-1 nouveau est également inséré, qui place le régime de fabrication des substances actives entrant dans la fabrication de médicaments à usage vétérinaire sous les mêmes contraintes.
L’article 11 modifie l’article L. 5138-4 du code de la santé publique afin d’exclure les excipients, compte tenu de leur spécificité, du champ de délivrance des certificats de respect des bonnes pratiques par l’ANSM dans le cadre de ses activités d’inspection. Il précise également en 2° que cette dernière coopère avec son équivalent européen, l’Agence européenne du médicament.
L’article 12 rétablit l’article L. 5138-5 dans le code de la santé publique. Les bonnes pratiques de l’Union européennes sont les normes minimales auxquelles doivent être soumises les substances actives importées de pays tiers.
Le 2° de l’article 14 prévoit que le rapport de synthèse que l’ANSM rend public chaque année doit notamment mettre l’accent sur « sur les actions entreprises dans le domaine de la prévention et de la répression de la falsification des médicaments ».
L’article 15 modifie l’article L. 5312-4 du code de la santé publique afin de préciser le rôle de veille et d’alerte de l’ANSM qui consiste à conduire des « mesures d’information appropriées », dans le but d’éviter que des médicaments soupçonnés d’être falsifiés ou d’être affectés de défauts de qualité ne soient mis à la disposition des patients. La personne en charge de la mise sur le marché d’un produit défaillant supportera, le cas échéant, le coût de ces mesures d’information.
L’article 16 prévoit, aux articles L. 5313-1 et L. 5313-3, que les inspections conduites par les inspecteurs de l’ANSM sont réalisées conformément aux bonnes pratiques définies par le directeur général de l’Agence.
Le I de l’article 17 précise les sanctions passibles en cas de manquement aux dispositions relatives à l’activité de courtage en produits pharmaceutiques. L’article L. 5421-12 rend passible de deux ans d’emprisonnement et de 30 000 euros d’amende le fait de réaliser cette activité sans déclaration auprès de l’agence. L’article L. 5421-13 prévoit que la fabrication, le courtage, la distribution, la vente ou encore la publicité de produits falsifiés sont punis de cinq ans d’emprisonnement et de 375 000 euros d’amende, peines pouvant être portées à sept ans d’emprisonnement et 750 000 euros d’amende en cas de circonstances aggravantes. Les personnes détentrices de médicaments falsifiés sans motif légitime sont également incriminables, conformément à l’article L. 5421-14, ainsi que la tentative de commettre ces mêmes délits, conformément à l’article L. 5421-15.
Le II de l’article 17 définit l’incrimination des manquements à la législation relative aux matières premières à usage pharmaceutique.
Il comporte sept articles nouveaux, à savoir :
– l’article L. 5438-1 qui décrit les manquements soumis à sanction financière, par exemple le fait de se soustraire au respect ou à la définition des bonnes pratiques, ou encore ne pas procéder aux audits ;
– l’article L. 5438-2 qui autorise l’agence nationale de sécurité des médicaments à prononcer des peines d’amende en cas d’infraction, l’article L. 5438-6 fixant les mêmes compétences en cas de tentatives ;
– l’article L. 5438-3 qui sanctionne le fait d’exercer une activité de fabrication, d’importation ou de distribution sans l’autorisation de l’agence nationale ;
– l’article L. 5438-4 qui incrimine la fabrication, le courtage, la distribution, la publicité, l’offre de vente, la vente, l’importation, l’exportation, l’achat de matières premières à usage pharmaceutique falsifiées. L’article L. 5438-5 punit quant à lui les détenteurs sans motif légitime de ces matières ;
– enfin, l’article L. 5438-7 qui permet d’engager la responsabilité des personnes morales déclarées pénalement responsables.
Les articles 19 et 20 de l’ordonnance modifient les articles L. 213-3 et L. 213-4 du code de la consommation en ôtant les références aux substances médicamenteuses : cette suppression est une conséquence des incriminations nouvelles définies par la même ordonnance. L’article 21 modifie en conséquence l’article L. 213-5 du code de la consommation.
L’article 22 porte sur l’application de l’ordonnance à Wallis-et-Futuna, territoire pour lequel quelques aménagements sont prévus.
3. Les modifications apportées par le présent article
Après une première ordonnance du juge des référés le 14 février 2013, le Conseil d’État, par décision n° 365317 du 17 juillet 2013 a invalidé une partie des dispositions de l’ordonnance en relevant que le droit européen « ne distingue, en vue de l’autorisation de la mise sur le marché des médicaments, que deux catégories de médicaments, correspondant à ceux soumis à prescription médicale et à ceux non soumis à prescription ».
En effet, les dispositions de la directive du 8 juin 2011 reprennent elles-mêmes une distinction résultant de la jurisprudence de la Cour de justice de l’Union européenne et notamment de son arrêt dit « DocMorris » du 11 décembre 2003, selon lequel les États membres ne peuvent exclure de la vente à distance au public au moyen de services de la société de l’information que les médicaments soumis à prescription.
Il en résulte donc que l’article L. 5125-34 du code de la santé publique selon lequel « seuls peuvent faire l’objet de l’activité de commerce électronique les médicaments de médication officinale qui peuvent être présentés en accès direct au public en officine » est contraire aux objectifs de la directive car il exclue de la possibilité de la vente en ligne les médicaments non soumis à prescription qui ne sont pas inscrits sur la liste de médication officinale.
Ainsi, le Conseil d’État a partiellement annulé l’article L. 5125-34 « en tant qu’il ne limite pas aux seuls médicaments soumis à prescription médicale obligatoire l’interdiction de faire l’objet de l’activité de commerce électronique ». Cette décision a eu pour effet de lever toute restriction à la vente des médicaments non soumis à prescription médicale obligatoire, qui peuvent donc actuellement être vendus en ligne sur des sites d’officines, conformément aux objectifs de la directive.
En conséquence, en II, l’alinéa 6 du présent article modifie l’article L. 5125-34 afin d’étendre la vente de médicaments en ligne à l’ensemble des « médicaments qui ne sont pas soumis à prescription obligatoire ». La liste des médicaments qui peuvent être vendus en ligne est désormais considérablement plus large que celles des 455 médicaments à prescription officinale initialement autorisés.
Outre cette mesure qui tire strictement les conséquences de la décision du Conseil d’État, le II du présent article apporte, aux alinéas 3 à 9 des modifications d’ordre rédactionnel ou de coordination à différentes dispositions figurant dans l’ordonnance ratifiée.
Mais l’alinéa 10 modifie entièrement la rédaction de l’article L. 5438-7 du code de la santé publique afin de préciser le régime de peines complémentaires encourues pour les infractions pénales décrites au chapitre visé : affichage ou diffusion de la décision de sanction, interdiction temporaire ou définitive d’exercer, confiscation de la chose qui a servi ou était destinée à commettre l’infraction ou la chose qui en est le produit. En conséquence, l’alinéa 16 fait figurer dans un nouvel article L. 5438-8 les dispositions permettant d’engager la responsabilité des personnes morales déclarées pénalement responsables, établies initialement par l’article 17 de l’ordonnance à l’article L. 5438-7, désormais modifié.
4. Les bonnes pratiques de dispensation des médicaments par voie électronique.
Conformément au troisième alinéa de l’article L. 5121-5 du code de la santé publique, établi par le 2° de l’article 3 de l’ordonnance, il est revenu à la ministre chargée de la santé de définir les bonnes pratiques de la dispensation de médicaments par voie électronique, par un arrêté du 20 juin 2013.
Un projet d’arrêté présenté au printemps 2013 visait à contraindre les pharmaciens de pratiquer les mêmes prix en ligne et en pharmacie et interdisait toute remise sur les frais de livraison : les médicaments vendus en ligne auraient donc été plus chers que ceux délivrés dans l’officine. Il était envisagé de rendre impossible la création de sites Internet proposant à la fois des médicaments et d’autres produits habituellement vendus par les pharmaciens. Des « newsletters » émanant du site auraient été interdites. Enfin les pharmaciens auraient eu l’obligation de disposer, en stock, dans les officines, de la totalité des produits mis en vente sur Internet.
En vertu de l’article L. 462-2 du code de commerce, l’Autorité de la concurrence est « obligatoirement consultée par le Gouvernement sur tout projet de texte réglementaire instituant un régime nouveau ayant directement pour effet : (…) 3° d’imposer des pratiques uniformes en matière de prix ou de conditions de vente ».
L’Autorité de la concurrence a rendu un avis défavorable (n° 13-A-12), le 10 avril 2013, selon lequel l’arrêté comporte « un ensemble important d’interdictions et de restrictions non justifiées par des considérations de santé publique » et « vise à limiter le développement de la vente en ligne de médicaments par les pharmaciens français, voire même à dissuader ces derniers d’utiliser ce canal de vente ». L’Autorité de la concurrence a ainsi rappelé son attachement à « la possibilité laissée aux pharmaciens de vendre en ligne des produits qui ne font pas partie du stock de leur officine et de contracter pour ce faire avec d’autres acteurs de la chaîne d’approvisionnement ».
L’arrêté du 20 juin 2013 relatif aux bonnes pratiques de dispensation des médicaments par voie électronique a, en conséquence, levé les restrictions litigieuses, en premier lieu en matière de prix et de modalités de stockage. Le site internet de vente en ligne de médicaments peut faire figurer d’autres produits vendus par le pharmacien mais doit comporter un onglet spécifique à la vente de médicaments. La distinction doit être claire par rapport aux autres produits.
Pour autant, l’arrêté encadre fortement la vente en ligne. À titre d’exemple la préparation des commandes ne peut se faire qu’au sein de l’officine, dans un espace adapté à cet effet. La sous-traitance à un tiers de tout ou partie de l’activité de vente par internet est interdite. Les liens hypertextes vers les sites des entreprises pharmaceutiques sont interdits, de même que la recherche de référencement dans des moteurs de recherche ou des comparateurs de prix contre rémunération. Les lettres d’information sont autorisées mais ne peuvent comporter, s’agissant du médicament, que des informations émanant des autorités sanitaires. L’hébergement des données de santé ne peut se faire qu’auprès d’hébergeurs agréés par le ministre chargé de la santé.
En matière de suivi et de conseil, l’arrêté prévoit que le site est conçu de sorte qu’aucun médicament ne puisse être vendu sans qu’un échange interactif pertinent ne soit rendu possible avant la validation de la commande. Une réponse automatisée à une question posée par le patient n’est pas suffisante pour assurer une information et un conseil adaptés au cas particulier du patient. Avant la validation de première commande, le pharmacien a la responsabilité de mettre en ligne un questionnaire dans lequel le patient doit renseigner son âge, son poids, son sexe, ses traitements en cours, ses antécédents allergiques et, le cas échéant, son état de grossesse ou d’allaitement, le patient attestant de la véracité de ces informations.
Au vu des exigences relevant du droit européen, votre rapporteur juge satisfaisantes les mesures d’encadrement de la vente en ligne des médicaments figurant tant dans le code de la santé publique que dans les actes réglementaires d’applications.
Il reste que l’ouverture de la vente en ligne aux médicaments, même non soumis à prescription médicale obligatoire, constitue un risque, le trafic des médicaments falsifiés passant essentiellement par la vente en ligne. Or ce risque n’est compensé que par un avantage minime pour le public, au regard de la densité et de la qualité du réseau français des pharmacies d’officines.
*
La Commission adopte l’article sans modification.
Article 6
(art. L. 5121-9-4 et L. 5124-6 du Code de la santé publique)
Information sur les motifs des décisions des exploitants de médicaments qui en suspendent ou arrêtent la commercialisation
Le présent définit de nouvelles obligations des titulaires d’autorisation de mise sur le marché de médicaments ainsi que des exploitants de ces médicaments prévues par la directive 2012/26/UE du 25 octobre 2012 qui a modifié, en ce qui concerne la pharmacovigilance, la directive 2001/83/CE du 6 novembre 2001 instituant un code communautaire relatif au médicament à usage humain.
1. La motivation des décisions de suspension ou d’arrêt de commercialisation par les titulaires d’autorisations de mise sur le marché
La directive 2012/26/UE modifie l’article 23 bis de la directive 2001/23/CE, qui prévoit que le titulaire de l’autorisation de mise sur le marché (AMM) prévient l’autorité compétente « si le médicament n’est plus mis sur le marché dans l’État membre concerné, de manière provisoire ou définitive ».
Le nouvel article 23 bis précise que le titulaire de l’AMM doit informer l’agence des raisons de son action. La procédure est décrite à l’article 123 qui oblige le titulaire de l’AMM à notifier aux États membres « toute action qu’il a engagée pour suspendre la mise sur le marché d’un médicament, retirer le médicament du marché, solliciter le retrait de l’autorisation de mise sur le marché ou ne pas en demander le renouvellement », dans n’importe quel État membre.
Le titulaire de l’AMM doit ainsi justifier son action. La directive 2012/26 l’oblige à se référer aux motifs exposés à l’article 116 ou au paragraphe 2 de l’article 117 qui recouvre les cas de figure suivants :
– le médicament est nocif ;
– le médicament ne permet pas d’obtenir de résultats thérapeutiques ;
– le rapport entre les bénéfices et les risques n’est pas favorable ;
– la spécialité n’a pas la composition qualitative et quantitative déclarée ;
– les contrôles sur le médicament ou sur les composants et les produits intermédiaires de la fabrication n’ont pas été effectués ou des exigences ou obligations relatives à l’octroi de l’autorisation de fabrication n’ont pas été respectées.
L’article 11 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé a fait figurer ces motifs de suspension ou de retrait d’une AMM aux 1° et 5° de l’article L. 5121-9 du code de la santé publique
La directive fixe donc pour objectif de contraindre le titulaire d’une AMM à se référer à ces motifs lorsqu’il informe les autorités nationales : cette modification représente bien une garantie supplémentaire en termes de sécurité sanitaire pour les patients. La nouvelle obligation de motivation imposée aux titulaires d’AMM garantit la qualité de la communication des informations nouvelles pouvant influencer l’évaluation bénéfices-risques.
Des sanctions pourront en effet être prises s’il s’avère que le retrait du produit a pour origine des événements survenus plusieurs mois voire plusieurs années avant la décision effective de retirer le produit du marché, alors que ces événements auraient dû être notifiés.
En conséquence, le II du présent article modifie l’article L. 5124-6 qui définit l’obligation pour le titulaire d’AMM d’informer l’ANSM de sa décision de suspendre ou de cesser la commercialisation d’un médicament. L’alinéa 5 du présent article prévoit que cette information est réalisée « de manière motivée ». L’alinéa 8 précise que la motivation se fonde sur l’un des motifs mentionnés aux 1° et 5° de l’article L. 5121-9, qui reprend les motifs listés à l’article 116 de la directive.
Le I du présent article modifie l’article L. 5121-9-4 qui, dans sa rédaction actuelle, prévoit que « le titulaire de l’autorisation de mise sur le marché qui arrête la commercialisation d’un médicament dans un autre État que la France alors que ce produit reste commercialisé en France doit en informer immédiatement l’Agence nationale de sécurité du médicament et des produits de santé et lui communiquer le motif de cet arrêt de commercialisation. »
L’alinéa 2 étend l’obligation d’information à « toute action engagée, en France ou dans un autre État membre, pour suspendre ou arrêter la commercialisation de ce médicament, pour solliciter le retrait de cette autorisation ou pour ne pas en demander le renouvellement ». Il prévoit également que le titulaire de l’AMM doit préciser si son action se fonde sur un des cas prévus aux 1° et 5° de l’article L. 5121-9.
Enfin, l’alinéa 3 instaure la même obligation d’information motivée lorsqu’une de ces actions est « engagée dans un pays tiers » : le titulaire de l’AMM doit alors en informer à la fois l’Agence nationale de sécurité du médicament et des produits de santé et l’Agence européenne des médicaments.
Votre rapporteur se félicite de cette amélioration de la qualité des informations transmises à l’ANSM, qui est un gage du renforcement de notre système de pharmacovigilance.
2. Un volet de transposition par voie réglementaire
Si l’ensemble des mesures prévues par l’article 6 du projet de loi seront d’application directe, votre rapporteur tient par ailleurs à souligner que des objectifs fixés par la directive 2012/26/UE du 25 octobre 2012 concernant la procédure d’urgence de l’Union européenne et les obligations des entreprises en matière d’importation et d’exportation vers des pays tiers de l’Union européenne viennent également d’être transposés dans le droit national par un décret n° 2013-923 du 16 octobre 2013.
La directive prévoit que la procédure d’urgence peut désormais être engagée soit par un État membre soit par la commission européenne sur la simple base d’inquiétudes résultant de l’évaluation des données issues des activités de pharmacovigilance.
Aux termes de l’article 107 decis, un État membre ou la Commission déclenche une procédure européenne d’évaluation lorsqu’une mesure d’urgence est jugée nécessaire à la suite de l’évaluation des données résultant des activités de pharmacovigilance, dans l’une des situations suivantes où il est envisagé de :
– suspendre ou retirer une autorisation de mise sur le marché ;
– interdire la délivrance d’un médicament ;
– refuser le renouvellement d’une autorisation de mise sur le marché ;
– signaler une nouvelle contre-indication.
L’Agence européenne est alors chargée de déterminer si le problème de sécurité touche tous les médicaments appartenant à la même gamme ou à la même classe thérapeutique, et d’entamer une évaluation scientifique. La Commission peut demander de prendre des mesures provisoires. La procédure mène à l’adoption de mesures harmonisées pour toute l’UE quant aux médicaments concernés.
Le décret n° 2013-923 modifie en ce sens les articles R. 5121-51-9 et R. 5121-157 du code de la santé publique.
La directive renforce d’autre part les obligations pesant sur les entreprises exportant des médicaments vers des pays tiers à l’Union européenne et sur les entreprises important des médicaments en provenance de ces pays, en précisant que ces médicaments ne peuvent être destinés ou obtenus que par des personnes autorisées et habilitées à recevoir les médicaments en vue de la distribution en gros dans ces pays. Le décret n° 2013-923 a modifié en ce sens l’article R. 5124-4 du code de la santé publique.
*
La Commission adopte l’amendement AS26 rédactionnel du rapporteur.
Puis elle adopte l’article 6 modifié.
Article 7
(art. L. 5121-1-2 et L. 5121-1-4 [nouveau] du code de la santé publique)
Harmonisation du contenu des prescriptions transfrontières
L’article L. 5111-2 du code de la santé publique définit les spécialités pharmaceutiques comme « tout médicament préparé à l’avance, présenté sous un conditionnement particulier et caractérisé par une dénomination spéciale ». Aux plans national comme international, des dénominations diverses sont utilisées pour désigner une spécialité pharmaceutique ou son principe actif, c’est-à-dire la substance responsable de l’action pharmacologique du médicament.
Cette variété rend malaisée l’identification des produits prescrits dans un État mais délivrés dans un autre. La directive 2011/24/UE du Parlement européen et du Conseil du 9 mars 2011 sur l’application des droits des patients en matière de soins de santé transfrontaliers prévoit que « lorsque des médicaments sont autorisés dans un État membre et ont été prescrits dans cet État membre pour un patient nommément désigné, il devrait, en principe, être possible de reconnaître ces prescriptions sur le plan médical et de délivrer les médicaments dans un autre État membre, dans lequel les médicaments sont autorisés ».
En conséquence, la directive d’exécution 2012/52 adoptée par la Commission européenne le 20 décembre 2012 fixe des règles communes relatives au contenu des prescriptions médicales destinées à être utilisées dans un autre État membre. Le présent article vise à compléter la transposition de ces règles.
1. La reconnaissance des prescriptions transfrontières est un gage d’accès aux soins transfrontaliers
Le droit d’accéder à des soins transfrontaliers au sein de l’Union européenne a été déduit de la libre-prestation de service, consacrée par le droit communautaire. L’article 56 du Traité sur le fonctionnement de l’Union européenne (TFUE) dispose en effet que « les restrictions à la libre prestation des services à l’intérieur de l’Union sont interdites à l’égard des ressortissants des États membres établis dans un État membre autre que celui du destinataire de la prestation ». Les services concernés sont définis par l’article 57 du TFUE comme « les prestations fournies normalement contre rémunération » qu’il s’agisse d’activités à caractère industriel, commercial, artisanal ou libéral.
La Cour de justice des Communautés européennes a affirmé à plusieurs reprises que la libre-prestation de services, régie auparavant par les articles 59 et 60 du traité CEE, s’applique aux soins de santé. À titre d’exemple, par l’arrêt Geraets-Smits et Peerboms du 12 juillet 2001, la Cour a déclaré que « les activités médicales relèvent du champ d’application de l’article 60 du traité ».
Dès lors, la jurisprudence de la CJCE a reconnu aux ressortissants communautaires le droit d’accéder à des soins dans un autre État membre tout en étant remboursés par leur État d’affiliation. Ce droit a été précisé par la directive 2011/24/UE du 9 mars 2011 relative à l’application des droits des patients en matière de soins de santé transfrontaliers qui instaure un système d’information sur les soins transfrontaliers et leur prise en charge reposant sur la création de points de contact nationaux. La directive cherche également à garantir la qualité et la sécurité de ces soins et à promouvoir la coopération entre États membres. À ce titre, elle consacre la reconnaissance des prescriptions médicales établies dans un autre État membre.
L’article 11 de la directive 2011/24/UE prévoit ainsi que les États veillent à la délivrance des prescriptions établies dans un autre État membre pour un médicament ou un dispositif médical autorisé à la vente sur leur territoire.
Afin de faciliter la reconnaissance de ces prescriptions, la Commission européenne a été chargée d’adopter un certain nombre de mesures afin de :
– permettre la vérification de l’authenticité de l’ordonnance par l’élaboration d’une liste d’éléments relatifs à l’identification du patient et du prescripteur devant figurer sur la prescription ;
– faciliter l’identification correcte des médicaments ou dispositifs médicaux prescrits ;
– renforcer l’intelligibilité des informations destinées aux patients concernant la prescription et l’utilisation du produit.
2. La coordination du contenu des prescriptions
En application de l’article 11 de la directive 2011/24, la Commission européenne a adopté, le 20 décembre 2012, la directive d’exécution 2012/52 établissant des mesures visant à faciliter la reconnaissance des prescriptions médicales établies dans un autre État membre. Elle établit la liste non exhaustive des éléments à inclure dans les prescriptions médicales établies à la demande d’un patient qui entend les utiliser dans un autre État membre. Il s’agit de :
– l’identification du patient : nom, prénom, date de naissance,
– l’authentification de la prescription indiquant sa date d’établissement,
– l’identification du professionnel de santé auteur de la prescription précisant ses nom, prénom, qualifications professionnelles, coordonnées directes, adresse professionnelle et signature,
– l’identification du produit prescrit avec la dénomination du produit, la forme pharmaceutique, la quantité, le dosage et la posologie.
En ce qui concerne la dénomination des médicaments prescrits, la directive impose de désigner les principes actifs par leur dénomination commune internationale (DCI), ou, à défaut, par leur dénomination commune usuelle. La dénomination commune internationale fait référence au nom non commercial du principe actif établi et recommandé par l’Organisation mondiale de la santé. Lorsque cette dernière n’a défini aucune dénomination commune internationale, le principe actif est désigné par une dénomination commune usuelle.
Afin d’améliorer la lisibilité de la prescription, la directive prévoit que pour les médicaments biologiques, outre la dénomination commune des principes actifs, le nom de marque, autrement dit le nom commercial déposé par le laboratoire, doit apparaître sur la prescription transfrontière.
L’annexe I de la directive 2001/83/CE définit le médicament biologique comme un produit dont le principe actif est une substance biologique c’est-à-dire « une substance qui est produite à partir d’une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité nécessitent une combinaison d’essais physico-chimico-biologiques, ainsi que la connaissance de son procédé de fabrication et de son contrôle. ».
Les médicaments biologiques tels que définis par le droit européen recouvrent ainsi :
– les médicaments immunologiques qui rassemblent les vaccins, sérums, toxines et allergènes ;
– les médicaments dérivés du sang et du plasma humains qui correspondent aux produits stables préparés à partir du sang humain ou de ses composants ;
– les médicaments de thérapie innovante qui peuvent être des médicaments issus de la thérapie génique (9), des médicaments issus de la thérapie cellulaire (10), des produits issus de l’ingénierie cellulaire (11) ou encore des médicaments combinés de thérapie innovante (12).
Enfin, la directive d’exécution 2012/52/UE prévoit que les points de contact nationaux établis par la directive 2011/2/UE prodiguent des informations sur les éléments devant apparaître sur leur prescription médicale aux patients désirant la faire exécuter dans un autre État membre. En France, le point de contact national est le Ministère de la santé.
3. Le droit français satisfait déjà en partie aux nouvelles exigences
La majeure partie des éléments devant figurer dans les prescriptions transfrontières en vertu de la directive d’exécution 2012/52 sont d’ores et déjà obligatoires pour toutes les prescriptions de médicament réalisées en France. En effet, l’article R. 5132-3 du code de la santé publique soumet la prescription de médicaments destinés à la médecine humaine à l’indication sur l’ordonnance de :
– l’identification complète du prescripteur comprenant son nom, sa qualité, son titre ou sa spécialité, son identifiant, son adresse, sa signature,
– la date de rédaction de l’ordonnance,
– l’identification du patient comprenant ses nom et prénoms, son sexe et son âge ainsi que, si nécessaire, sa taille et son poids,
– l’identification du produit prescrit (nom et dosage) et notamment sa posologie et son mode d’emploi,
– la durée du traitement ou le nombre d’unités de conditionnement et, si nécessaire, le nombre de renouvellements de la prescription,
– l’éventuelle mention manuscrite « non substituable » par des médicaments génériques,
– la nature et la périodicité des examens prescrits pour les médicaments nécessitant une surveillance particulière pendant le traitement,
– la mention garantissant que le prescripteur a informé le patient des risques encourus pour les médicaments soumis à prescription restreinte.
Cependant, l’utilisation de la dénomination commune des principes actifs dans les prescriptions médicales n’est pas encore effective en France, malgré une intervention récente du législateur.
L’article R. 5132-3 du code de la santé publique dispose en effet que les prescriptions médicales comprennent « la dénomination du médicament […] ou le principe actif du médicament désigné par sa dénomination commune ». Les prescripteurs ne sont donc pas tenus, en principe, d’utiliser sur l’ordonnance la DCI ou la dénomination commune usuelle d’une spécialité pharmaceutique. En pratique, on observe d’ailleurs que les médecins recourent majoritairement au nom de marque pour établir leurs prescriptions.
Pourtant la dénomination commune internationale permet d’identifier facilement les principes actifs d’un médicament, ainsi que les fausses innovations thérapeutiques. Ainsi, le principe actif du Mediator, le benfluorex, s’il avait été inscrit sur les ordonnances, aurait peut-être permis aux professionnels et aux patients de reconnaître le caractère anorexigène de ce médicament.
Néanmoins, l’article 50 de la loi n° 2008-1330 du 17 décembre 2008 de financement de la sécurité sociale pour 2009 a rendu obligatoire la prescription en dénomination commune pour les spécialités figurant dans un groupe générique.
Surtout, la généralisation de la prescription en dénomination commune a été prévue par l’article 19 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
Cette loi a instauré un nouvel article L. 5121-1-2 dans le code de la santé publique afin de rendre obligatoire, dans la prescription d’une spécialité pharmaceutique, la mention de ses principes actifs, désignés par leur dénomination commune internationale ou, à défaut, par leur dénomination dans la pharmacopée européenne ou française (13). S’il n’existe pas de DCI, la dénomination commune usuelle des principes actifs doit apparaître sur la prescription médicale. La mention de la dénomination de fantaisie de la spécialité, autrement dit de son nom de marque, est facultative et ne permet pas à elle seule la délivrance du produit.
Or l’article L. 5121-1-2 du code de la santé publique n’entrera en vigueur que lors de la publication de décrets d’application relatifs à la certification des logiciels d’aide à la prescription, et à la constitution d’une base de données publique sur les produits de santé, et au plus tard le 1er janvier 2015.
Dès lors il est possible que l’obligation d’utilisation de la dénomination commune des principes actifs entre en vigueur en France plusieurs mois après la date limite de transposition de la directive d’exécution 2012/52/UE, fixée au 25 octobre 2013.
Mais l’adoption de ces textes requiert la certification par la Haute autorité de santé (HAS) des logiciels d’aide à la dispensation en dénomination commune internationale, prévue par l’article L. 161-38 du code de la santé publique.
Cet article prévoit que la HAS « garantit que ces logiciels assurent la traduction des principes actifs des médicaments selon leur dénomination commune internationale recommandée par l’Organisation mondiale de la santé ou, à défaut, leur dénomination dans la pharmacopée européenne ou française ».
Votre rapporteur insiste donc sur la nécessité d’une parution rapide de ces décrets.
4. Les modifications apportées en matière de dénomination des spécialités pharmaceutiques
L’obligation de mention du nom de marque pour les prescriptions transfrontières de médicaments biologiques n’existe pas en droit français et nécessite par conséquent une modification du code de la santé publique.
En conséquence, le II du présent article insère dans le code de la santé publique un article L. 5121-1-4 qui rend obligatoire, lorsqu’une prescription est « établie à la demande d’un patient en vue de l’utiliser dans un autre État membre de l’Union européenne » :
– la mention des principes actifs du produit prescrit par utilisation de la dénomination commune internationale ou, à défaut, de la dénomination de la pharmacopée,
– l’utilisation du nom de marque de la spécialité pharmaceutique.
Cette obligation de double mention du principe actif et de la spécialité ne concerne que les médicaments biologiques tels que définis par le droit européen, qui recouvrent à la fois :
– les médicaments immunologiques mentionnés au 6° de l’article L. 5121-1,
– les médicaments biologiques au sens de la législation française, au 14° de l’article L. 5121-1, soit « tout médicament dont la substance active est produite à partir d’une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité nécessitent une combinaison d’essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de son contrôle ». Ils correspondent aux médicaments biotechnologiques au sens européen,
– les médicaments biologiques similaires mentionnés au 15° de l’article L. 5121-1,
– les médicaments dérivés du sang, mentionnés à l’article L. 5121-3,
– les médicaments de thérapie innovante mentionnés au point a du 1 de l’article 2 du règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007, qui recouvrent les médicaments de thérapie génique, de thérapie cellulaire somatique ou produits issus de l’ingénierie tissulaire, ainsi que les médicaments combinés de thérapie innovante mentionnés au point d du 1 de l’article 2 du même règlement.
Enfin, à des fins de coordination avec le nouvel article L. 5121-1-4, le I du présent article modifie l’article L. 5121-1-2 relatif à l’obligation de prescription en dénomination commune internationale, et qui n’est pas encore entré en vigueur.
*
La Commission adopte l’amendement AS29 rédactionnel du rapporteur.
Elle examine ensuite l’amendement AS 10 du rapporteur.
M. le rapporteur. Cet amendement prévoit l’attribution d’un label « éthique » symbolisé par un pictogramme distinctif apposé sur les médicaments dérivés du sang produits dans des conditions éthiques au sens de la législation française, c’est-à-dire à partir de dons bénévoles, gratuits et anonymes. Je rappelle que 1,7 million de donneurs se mobilisent en la matière ainsi que des milliers de salariés de la filière du sang. Il en résulte que la France est autosuffisante pour tous les produits dérivés du sang.
Une barrière avait été instaurée par la législation pour éviter que soient importés des médicaments non produits dans ces conditions, au travers d’une AMM délivrée par l’ANSM. Mais depuis l’AMM centralisée européenne, elle a volé en éclat et 40 % des médicaments dérivés du sang vendus dans les hôpitaux français ne répondent pas à ces règles éthiques.
J’espère que ce label conduira à une prise de conscience de la chance que nous avons en France d’avoir une filière reposant sur de telles règles et qu’il pourra donner lieu demain à un label européen. D’ailleurs, cette mesure est conforme au droit européen : plusieurs décisions de la Commission européenne vont dans ce sens. Quant au traité sur le fonctionnement de l’Union européenne (TFUE), il prévoit que les États membres peuvent modifier la circulation des produits en tenant compte de la qualité et de la sécurité lorsqu’il s’agit de médicaments dérivés du sang.
M. Jean-Pierre Barbier. Je m’associe aux propos du rapporteur. Mais quand il s’agira pour les hôpitaux d’acheter des produits dérivés du sang, ils procéderont par appel d’offre, et comme le critère du prix est essentiel, ils risquent de les acquérir à l’étranger, en dehors de la filière éthique du sang. Quel impact aura sur les patients le fait de recevoir des produits ne disposant pas du label ? Ne risque-t-on pas de susciter chez eux une inquiétude ?
M. le rapporteur. Il y a en effet une guerre des prix dans ce domaine : des centrales d’achat achètent en grande quantité des produits dérivés du sang et coupleront dans les prochains appels d’offre les achats d’immunoglobulines et d’albumine de manière à réduire les écarts de prix. En outre, une disposition du PLFSS, votée à l’unanimité, prévoit, par une fiscalité différenciée, de baisser le coût de production des médicaments produits dans des conditions éthiques – ce qui permettra également de réduire ces écarts.
Je rappelle que, depuis un certain nombre d’années, lorsque nous ne sommes pas capables de fournir aux malades français des médicaments dérivés du sang produits dans des conditions éthiques, une AMM dérogatoire de deux ans délivrée par l’ANSM permet de commercialiser en France des médicaments produits aux États-Unis dans d’autres conditions – je pense notamment aux immunoglobulines anti-D perfusées aux femmes enceintes et comportant une allo-immunisation materno-fœtale, qui ne sont fabriquées que dans ce pays. Il serait d’ailleurs souhaitable d’éviter ce type de monopole car le jour où le laboratoire aura un problème avec son usine, on pourra avoir une catastrophe sanitaire.
Si l’on accepte une telle commercialisation, c’est qu’il n’y pas de risque excessif sur les produits sanguins labiles, c’est-à-dire les perfusions de globules rouges ou blancs. Il est reconnu partout dans le monde que la gratuité du don participe de la sécurité. C’est la raison pour laquelle la Food and Drug Administration (FDA) – l’Agence américaine des produits alimentaires et médicamenteux – labellise elle-même ses propres produits sanguins en fonction de leur origine éthique.
Les médicaments dérivés du sang seront maintenus en quarantaine 60 jours avant d’être traités et commercialisés, et ils feront l’objet de prélèvements sérologiques. Si l’Organisation mondiale de la santé (OMS) considère que la gratuité participe toujours de la sécurité, le phénomène sera marginal. Il n’y a pas d’inquiétude à avoir pour la sécurité des patients.
Par contre, nous devons revendiquer, au travers de ce label, l’héritage légué des suites de la Seconde Guerre mondiale du système de dons éthiques, qui fonctionne dans un marché mondialisé.
M. Gérard Sebaoun. Je suis en faveur de cet amendement. Mais certains patients ne risquent-ils pas de demander impérativement à bénéficier de produits éthiques ?
Mme la présidente Catherine Lemorton. J’ai les mêmes réticences s’agissant du marquage de fabrication française sur les médicaments.
M. le rapporteur. Quand j’ai auditionné les associations d’usagers qui reçoivent des immunoglobulines en perfusion, certains disent qu’ils sont très favorables à un système éthique, d’autres que ce qui compte pour eux est d’avoir le médicament le plus innovant, offrant la meilleure tolérance possible.
C’est la raison pour laquelle, dans le rapport sur la filière du sang, je propose, à côté des trois principes fondateurs de celle-ci que sont la sécurité, l’autosuffisance et l’éthique, d’en introduire un quatrième, qui est celui de la pluralité. Cela signifie que soient systématiquement garantis, dans tous les établissements français, la continuité de traitement aux patients et le choix des médicaments, de façon à ce que les malades puissent bénéficier du traitement le plus efficace et pour lequel ils auront la meilleure tolérance – ce qui n’est pas le cas aujourd’hui.
Enfin, si les patients exigent demain d’avoir un produit éthique, cela contrebalancera le critère du prix.
La Commission adopte l’amendement.
Elle en vient à l’amendement AS2 du rapporteur.
M. le rapporteur. Il s’agit d’autoriser le Gouvernement à déterminer par décret la liste des caractéristiques à préciser lors de la prescription d’un dispositif médical. En effet, la directive d’exécution de la Commission européenne du 20 décembre 2012 prévoit une prescription transfrontalière des dispositifs médicaux et il n’y a pas de texte à cet effet dans le droit français.
La Commission adopte l’amendement.
Elle adopte ensuite l’article 7 modifié.
Puis elle adopte à l’unanimité l’ensemble du projet de loi modifié.
___
Dispositions en vigueur ___ |
Texte du projet de loi ___ |
Texte adopté par la Commission ___ |
Projet de loi portant diverses dispositions d’adaptation au droit de l’Union européenne dans le domaine de la santé |
Projet de loi portant diverses dispositions d’adaptation au droit de l’Union européenne dans le domaine de la santé | |
Article 1er |
Article 1er | |
I. – Hors le cas où leur responsabilité est encourue en raison d’un défaut d’un produit de santé, les professionnels autorisés à user du titre d’ostéopathe ou de chiropracteur ne sont responsables des conséquences domma-geables d’actes accomplis dans le cadre de leur activité professionnelle qu’en cas de faute. |
I. – Hors … … raison du défaut … … faute. Amendement AS12 | |
II. – Les professionnels autorisés à user du titre d’ostéopathe ou de chiropracteur et exerçant leur activité à titre libéral sont tenus de souscrire une assurance destinée à les garantir pour leur responsabilité civile susceptible d’être engagée en raison de dommages subis par des tiers et résultant d’atteintes à la personne, survenant dans le cadre de l’ensemble de cette activité. |
||
Les contrats d’assurance souscrits en application de l’alinéa précédent peuvent prévoir des plafonds de garantie. Le montant minimal de ces plafonds est fixé par décret en Conseil d’État. |
||
Les dispositions prévues aux articles L. 251-2 et L. 251-3 du code des assurances relatives aux contrats d’assurance souscrits par les professionnels de santé en application de l’article L. 1142-2 du code de la santé publique sont applicables aux contrats d’assurance souscrits par les professionnels autorisés à user du titre d’ostéopathe ou de chiropracteur. |
||
Au 1er janvier 2014, tout profes-sionnel autorisé à user du titre d’ostéopathe ou de chiropracteur doit être en mesure de justifier que sa responsabilité est couverte dans les conditions prévues au présent article. |
Au 1er janvier 2015, tout … … article. Amendement AS1 | |
Article 2 |
Article 2 | |
Le manquement à l’obligation d’assurance prévue à l’article 1er de la présente loi est puni de 45 000 € d’amende. |
(Sans modification) | |
Les personnes physiques coupables de l’infraction mentionnée au présent article encourent également la peine complémentaire d’interdiction, selon les modalités prévues par l’article 131-27 du code pénal, d’exercer l’activité professionnelle ou sociale dans l’exercice de laquelle ou à l’occasion de l’exercice de laquelle l’infraction a été commise. Cette interdiction est portée à la connaissance du directeur général de l’agence régionale de santé. |
||
Article 3 |
Article 3 | |
Code de la santé publique |
I. – Le chapitre Ier du titre III du livre Ier de la cinquième partie du code de la santé publique est ainsi modifié : |
|
Art. L. 5131-1. – On entend par produit cosmétique toute substance ou mélange destiné à être mis en contact avec les diverses parties superficielles du corps humain, notamment l’épiderme, les systèmes pileux et capillaire, les ongles, les lèvres et les organes génitaux externes, ou avec les dents et les muqueuses buccales, en vue, exclusivement ou principalement, de les nettoyer, de les parfumer, d’en modifier l’aspect, de les protéger, de les maintenir en bon état ou de corriger les odeurs corporelles. |
1° À l’article L. 5131-1, le mot : « diverses » est supprimé et les mots : « , notamment l’épiderme, les systèmes pileux et capillaire, les ongles, les lèvres et les organes génitaux externes, » sont remplacés par les mots : « (l’épiderme, les systèmes pileux et capillaire, les ongles, les lèvres et les organes génitaux externes) » ; |
|
2° L’article L. 5131-2 est ainsi modifié : |
||
Art. L. 5131-2. – L’ouverture et l’exploitation de tout établissement de fabrication, de conditionnement ou d’importation, même à titre accessoire, de produits cosmétiques, de même que l’extension de l’activité d’un établissement à de telles opérations, sont subordonnées à une déclaration auprès de l’Agence nationale de sécurité du médicament et des produits de santé . |
a) Au premier alinéa, les mots : « , de conditionnement ou d’impor-tation » sont remplacés par les mots : « ou de conditionnement » ; |
|
b) Les deuxième, troisième et quatrième alinéas sont remplacés par deux alinéas ainsi rédigés : |
||
Cette déclaration est effectuée par le fabricant, ou par son représentant ou par la personne pour le compte de laquelle les produits cosmétiques sont fabriqués, ou par le responsable de la mise sur le marché des produits cosmétiques importés pour la première fois d’un État non membre de l’Union européenne ou non partie à l’accord sur l’Espace économique européen. Elle indique les personnes qualifiées responsables désignées en application du quatrième alinéa. |
||
Toute modification des éléments figurant dans la déclaration initiale doit faire l’objet d’une nouvelle déclaration dans les mêmes formes. |
« Toute modification des éléments constitutifs de la déclaration est communiquée à l’agence. |
|
La personne qui dirige un établissement mentionné au premier alinéa désigne une ou plusieurs personnes qualifiées responsables de la fabrication, du conditionnement, de l’importation, des contrôles de qualité, de l’évaluation de la sécurité pour la santé humaine, de la détention et de la surveillance des stocks de matières premières et de produits finis. Ces personnes doivent posséder des connaissances scientifiques suffisantes attestées par des diplômes, titres ou certificats figurant sur une liste établie par arrêté des ministres chargés de l’artisanat, de l’enseignement supérieur, de l’industrie et de la santé ou justifier d’une expérience pratique appropriée dont la durée et le contenu sont déterminés dans les mêmes conditions. |
« Les personnes qualifiées en charge de l’évaluation de la sécurité doivent posséder une formation universitaire telle que mentionnée à l’article 10 du règlement (CE) n° 1223/2009 du 30 novembre 2009 ou une formation équivalente figurant sur une liste établie par arrêté des ministres chargés de la santé, de l’industrie et de l’enseignement supérieur. » ; |
« Les … … supérieur ou une formation reconnue équivalente par un État membre de l’Union européenne. » ; Amendement AS9 |
3° Les articles L. 5131-3 à L. 5131-11 sont remplacés par les dispositions suivantes : |
||
Art. L. 5131-3. – Les dispositions de l’article L. 5131-2 ne s’appliquent pas aux établissements qui importent des produits cosmétiques en provenance exclusivement d’Etats membres de l’Union européenne ou parties à l’accord sur l’Espace économique européen. |
« Art. L. 5131-3. – Les produits cosmétiques mis à disposition sur le marché satisfont aux dispositions du règlement (CE) n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques. |
|
« L’autorité compétente men-tionnée au paragraphe 5 de l’arti-cle 6, au paragraphe 3 de l’article 11, au paragraphe 5 de l’article 13 et aux articles 23, 24, 25, 26, 27, 28, 29 et 30 du règlement mentionné ci-dessus est l’Agence nationale de sécurité du médicament et des produits de santé. Le ministre chargé de la consommation et les agents mentionnés au 1° de l’article L. 215-1 du code de la consommation ont également la qualité d’autorité compétente pour la mise en œuvre du paragraphe 5 de l’article 6, du para-graphe 3 de l’article 11, du paragraphe 5 de l’article 13, du paragraphe 5 de l’article 23 et des articles 24, 25, 26, 28, 29 et 30 du règlement, dans la limite des pouvoirs dont ils disposent en vertu des dispositions du code de la consom-mation et du présent code. |
||
Art. L. 5131-4. – Les produits cosmétiques mis sur le marché ne doivent pas nuire à la santé humaine lorsqu’ils sont appliqués dans les conditions normales ou raisonnablement prévisibles d’utilisation compte tenu, notamment, de la présentation du produit, des mentions portées sur l’étiquetage ainsi que de toutes autres informations destinées aux consommateurs. |
« Art. L. 5131-4. – L’Agence nationale de sécurité du médicament et des produits de santé publie les principes de bonnes pratiques de laboratoire applicables aux études de sécurité non cliniques destinées à évaluer la sécurité des produits cosmétiques pour la mise en œuvre de l’article 10 du règlement (CE) n° 1223/2009 du 30 novembre 2009, ainsi que les règles applicables à l’inspection et à la vérification des bonnes pratiques de laboratoire. Elle définit les règles relatives à la délivrance des documents attestant le respect de ces bonnes pratiques. |
|
Art. L. 5131-5. – La fabrication des produits cosmétiques doit être réalisée en conformité avec les bonnes pratiques de fabrication dont les principes sont définis par décision de l’Agence nationale de sécurité du médicament et des produits de santé . L’évaluation de la sécurité pour la santé humaine de ces produits doit être exécutée en conformité avec les bonnes pratiques de laboratoire dont les principes sont définis dans les mêmes conditions. Les règles générales relatives aux modalités d’inspection et de vérification des bonnes pratiques de laboratoire pour les produits cosmétiques ainsi qu’à la délivrance de documents attestant de leur respect sont définies par décision de l’Agence nationale de sécurité du médicament et des produits de santé . |
« Art. L. 5131-5. – I. – Toute personne responsable et tout distributeur de produits cosmétiques peuvent déclarer, en complément de leurs obligations découlant de l’article 23 du règlement (CE) n° 1223/2009 du 30 novembre 2009 à l’Agence nationale de sécurité du médicament et des produits de santé, les effets indésirables qui, bien que n’ayant pas le caractère d’effets indésirables graves au sens du point p du paragraphe 1 de l’article 2 de ce règlement, leur paraissent revêtir un caractère de gravité justifiant une telle déclaration. |
« Art. L. 5131-5. – … … 2009, les autres effets indésirables à l’Agence nationale de sécurité du médicament et des produits de santé. Amendement AS5 |
« II. – Tout professionnel de santé ayant connaissance d’un effet indésirable grave au sens du point p du paragraphe 1 de l’article 2 du règlement (CE) n° 1223/2009 du 30 novembre 2009, susceptible de résulter de l’utilisation d’un produit cosmétique, le déclare sans délai à l’Agence nationale de sécurité du médicament et des produits de santé. Il peut déclarer, en outre, les autres effets indésirables dont il a connaissance. Dans sa déclaration, il fait état, le cas échéant, d’un mésusage. |
« II. – … … connaissance. Il peut, d’autre part, déclarer les effets susceptibles de résulter d’un mésusage. Amendement AS6 | |
« Tout utilisateur professionnel peut procéder à la déclaration d’effets indésirables à l’Agence nationale de sécurité du médicament et des produits de santé. |
« Tout … … santé. Il peut, d’autre part, déclarer les effets susceptibles de résulter d’un mésusage. Amendement AS7 | |
« Tout consommateur de produits cosmétiques peut procéder à la déclaration d’effets indésirables à l’Agence nationale de sécurité du médicament et des produits de santé, en faisant état, le cas échéant, d’un mésusage. |
« Tout … … santé. Il peut, d’autre part, déclarer les effets susceptibles de résulter d’un mésusage. Amendement AS8 | |
Art. L. 5131-6. – Un produit cosmétique ne peut être mis sur le marché à titre gratuit ou onéreux que : – si son récipient et son emballage comportent le nom ou la raison sociale et l’adresse du fabricant ou du responsable de la mise sur le marché, établi dans un État membre de l’Union européenne ou partie à l’accord sur l’Espace économique européen, ainsi que les autres mentions prévues par le décret mentionné au 1° de l’article L. 5131-11 ; en cas de pluralité d’adresses, celle qui est soulignée désigne le lieu de détention du dossier prévu à l’alinéa suivant ; |
« Art. L. 5131-6. – En cas de doute sérieux quant à la sécurité d’une substance entrant dans la composition d’un produit cosmétique, l’Agence nationale de sécurité du médicament et des produits de santé peut mettre en demeure la personne responsable de ce produit de lui communiquer les informations mention-nées à l’article 24 du règlement (CE) n° 1223/2009 du 30 novembre 2009. Cette mise en demeure peut être assortie d’une astreinte au plus égale à 500 € par jour de retard à compter de la date fixée par l’agence. Le montant maximal de l’astreinte mise en recouvrement ne peut être supérieur au montant maximal de l’amende prévue à l’article L. 5431-9. |
|
– et si le fabricant, ou son représentant, ou la personne pour le compte de laquelle le produit cosmétique est fabriqué, ou le responsable de la mise sur le marché d’un produit cosmétique importé pour la première fois d’un Etat non membre de l’Union européenne ou non partie à l’accord sur l’Espace économique européen tient effectivement à la disposition des autorités de contrôle, à l’adresse mentionnée ci-dessus, un dossier rassemblant toutes informations utiles au regard des dispositions des articles L. 5131-4 et L. 5131-5, notamment sur la formule qualitative et quantitative, les spécifications physico-chimiques et microbiologiques, les conditions de fabrication et de contrôle, l’évaluation de la sécurité pour la santé humaine, les effets indésirables de ce produit cosmétique, et les preuves de ses effets revendiqués lorsque la nature de l’effet ou du produit le justifie. L’obligation d’indiquer dans le dossier la formule du produit ne s’applique pas aux parfums proprement dits ni aux compositions parfumantes pour lesquels les informations sont limitées au numéro de code de la composition parfumante et à l’identité de son fournisseur. |
||
Art. L. 5131-7. – La mise sur le marché à titre gratuit ou onéreux d’un produit cosmétique est subordonnée à la transmission aux centres antipoison mentionnés à l’article L. 6141-4, désignés par arrêté des ministres chargés de la consommation, de l’industrie et de la santé, d’informations adéquates et suffisantes concernant les substances utilisées dans ce produit. |
« Art. L. 5131-7. – Pour tout produit cosmétique mis sur le marché ou importé pour la première fois d’un État non membre de l’Union européenne ou non partie à l’accord sur l’Espace économique européen avant le 11 juillet 2013, le fabricant, son représentant, la personne pour le compte de laquelle le produit est fabriqué ou, en cas d’importation, le responsable de la mise sur le marché conserve, jusqu’au 11 juillet 2020, le dossier rassemblant les informations sur le produit. |
|
La liste de ces informations est fixée par arrêté des ministres chargés de la consommation, de l’industrie et de la santé. |
« Les centres antipoison men-tionnés à l’article L. 6141-4 conservent jusqu’au 11 juillet 2020 les informations adéquates et suffisantes, reçues avant le 11 juillet 2013, qui concernent les substances utilisées dans les produits cosmétiques. |
« Les … … 2013, concernant les substances … … cosmétiques. Amendement AS13 |
Art. L. 5131-7-1. – Sans préju-dice des protections dont le produit peut faire l’objet, notamment au titre du secret commercial et des droits de propriété intellectuelle, le fabricant ou son mandataire ou la personne pour le compte de laquelle le produit cosmétique est fabriqué ou le responsable de la mise sur le marché du produit cosmétique met à la disposition du public, par des moyens appropriés, y compris des moyens électroniques : 1° La formule qualitative du produit cosmétique ; en ce qui concerne les compositions parfumantes et les parfums, ces informations sont limitées à leur nom, à leurs numéros de code et à l’identité de leur fournisseur ; 2° Les quantités de substances qui entrent dans la composition de ce produit et répondent aux critères d’une des classes ou catégories de danger suivantes, visées à l’annexe I du règlement (CE) n° 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l’étiquetage et à l’emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) n° 1907/2006 : a) Les classes de danger 2.1 à 2.4, 2.6 et 2.7, 2.8 types A et B, 2.9, 2.10, 2.12, 2.13 catégories 1 et 2, 2.14 catégories 1 et 2, 2.15 types A à F ; b) Les classes de danger 3.1 à 3.6, 3.7 effets néfastes sur la fonction sexuelle et la fertilité ou sur le développement, 3.8 effets autres que des effets narcotiques, 3.9 et 3.10 ; c) La classe de danger 4.1 ; d) La classe de danger 5.1. ; 3° Les données existantes en matière d’effets indésirables pour la santé humaine résultant de son utilisation. |
Abrogé |
|
Art. L. 5131-7-2. – Sans préju-dice des obligations générales découlant de l’article L. 5131-4, il est interdit de : a) Mettre sur le marché des produits cosmétiques dont la formulation finale, afin de satisfaire aux exigences du présent chapitre, a fait l’objet d’une expérimentation animale au moyen d’une méthode autre qu’une méthode alternative. Le cas échéant, les méthodes alternatives validées et adoptées par la Commission européenne sont fixées par arrêté du ministre chargé de la santé après avis de l’Agence nationale de sécurité du médicament et des produits de santé ; b) Mettre sur le marché des produits cosmétiques contenant des ingrédients ou des combinaisons d’ingrédients qui, afin de satisfaire aux exigences du présent chapitre, ont fait l’objet d’une expérimentation animale au moyen d’une méthode autre qu’une méthode alternative. Le cas échéant, les méthodes alternatives validées et adoptées par la Commission européenne sont fixées par l’arrêté mentionné au a ; c) Réaliser, afin de satisfaire aux exigences du présent chapitre, des expérimentations animales portant sur des produits cosmétiques finis ; d) Réaliser, afin de satisfaire aux exigences du présent chapitre, des expérimentations animales portant sur des ingrédients ou des combinaisons d’ingrédients. Les méthodes alternatives validées sont précisées dans le règlement (CE) n° 440/2008 de la Commission du 30 mai 2008 établissant des méthodes d’essai conformément au règlement (CE) n° 1907/2006 du Parlement européen et du Conseil concernant l’enregistrement, l’évalua-tion et l’autorisation des substances chimiques ainsi que les restrictions applicables à ces substances (REACH) ou dans l’arrêté mentionné aux a et b. Ces méthodes alternatives sont décrites dans un arrêté des ministres chargés de la santé, de la consommation et de l’industrie, pris sur proposition de l’Agence nationale de sécurité du médicament et des produits de santé . L’interdiction mentionnée aux a et b ci-dessus entre en vigueur au plus tard le 11 mars 2013 pour les expérimentations concernant la toxicité des doses répétées, la toxicité pour la reproduction et la toxicocinétique. |
Abrogé |
|
Art. L. 5131-7-3 Dans des circonstances exceptionnelles, lorsque la sécurité d’un ingrédient existant de produit cosmétique suscite de graves préoccupations, l’Agence nationale de sécurité du médicament et des produits de santé peut demander à la Commission européenne d’accorder une dérogation aux dispositions de l’article L. 5131-7-2. Cette demande comporte une évaluation de la situation et indique les mesures dérogatoires jugées nécessaires.. – |
Abrogé |
|
Art. L. 5131-8. – Toute personne ayant accès au dossier et aux informations mentionnés aux articles L. 5131-6 et L. 5131-7 est tenue au secret professionnel dans les conditions prévues aux articles 226-13 et 226-14 du code pénal. |
« Art. L. 5131-8. – Un décret en Conseil d’État détermine les modalités d’application du présent chapitre, notamment : « 1° Les modalités de présen-tation et le contenu de la déclaration prévue à l’article L. 5131-2 ; |
|
« 2° Les modalités d’étiquetage des produits cosmétiques mentionnés au paragraphe 4 de l’article 19 du règlement (CE) n° 1223/2009 du 30 novembre 2009, après avis du Conseil national de la consommation ; |
||
« 3° Les modalités de mise en œuvre du système de cosmétovigilance prévu à l’article L. 5131-5. » |
||
Art. L. 5131-9. – I. – Pour l’application du présent article, on entend par effet indésirable grave une réaction nocive et non recherchée, se produisant dans les conditions normales d’emploi d’un produit cosmétique chez l’homme ou résultant d’un mésusage qui, soit justifierait une hospitalisation, soit entraînerait une incapacité fonctionnelle permanente ou temporaire, une invalidité, une mise en jeu du pronostic vital immédiat, un décès ou une anomalie ou une malformation congénitale. Pour la mise en oeuvre du système de cosmétovigilance, tout professionnel de santé ayant constaté un effet indésirable grave susceptible d’être dû à un produit cosmétique mentionné à l’article L. 5131-1 doit en faire la déclaration sans délai au directeur général de l’Agence nationale de sécurité du médicament et des produits de santé . Ce professionnel déclare en outre les effets indésirables qui, bien que ne répondant pas à la définition mentionnée ci-dessus, lui paraissent revêtir un caractère de gravité justifiant une telle déclaration. Dans sa déclaration, le professionnel de santé précise notamment si l’effet indésirable résulte d’un mésusage. II. – Les fabricants, ou leurs représentants, ou les personnes pour le compte desquelles les produits cosmétiques sont fabriqués, ou les responsables de la mise sur le marché des produits cosmétiques importés pour la première fois d’un Etat non membre de l’Union européenne ou non partie à l’accord sur l’Espace économique européen, ou les distributeurs, sont tenus de participer au système national de cosmétovigilance. Cette obligation est réputée remplie par la mise en oeuvre des dispositions de l’article L. 221-1-3 du code de la consommation. L’Agence française de sécurité sanitaire des produits de santé est tenue informée par les autorités administratives compé-tentes mentionnées à l’article L. 221-1-3 du code de la consommation. |
Abrogé |
|
Art. L. 5131-10. – Les fabricants, ou leurs représentants, ou les personnes pour le compte desquelles les produits cosmétiques sont fabriqués, ou les responsables de la mise sur le marché des produits cosmétiques importés pour la première fois d’un Etat non membre de l’Union européenne ou non partie à l’accord sur l’Espace économique européen sont tenus, en cas de doute sérieux sur l’innocuité d’une ou de plusieurs substances, de fournir au directeur général de l’Agence nationale de sécurité du médicament et des produits de santé lorsqu’il leur en fait la demande motivée, la liste de leurs produits cosmétiques dans la composition desquels entrent une ou plusieurs substances désignées par lui ainsi que la quantité de ladite substance présente dans le produit. L’agence prend toutes mesures pour protéger la confidentialité des informations qui lui sont transmises au titre de l’alinéa précédent. |
Abrogé |
|
Art. L. 5131-11. – Les modalités d’application du présent chapitre sont déterminées par décret en Conseil d’Etat et notamment : 1° Après avis du Conseil national de la consommation, les règles auxquelles doivent satisfaire les récipients et emballages des produits cosmétiques afin que soient lisibles et indélébiles le nom ou la raison sociale ainsi que l’adresse du fabricant ou du responsable de la mise sur le marché, le contenu nominal du produit ; sa date de durabilité minimale, les précautions d’emploi, la numérotation des lots de fabrication ou la référence permettant l’identification de la fabrication ; la fonction du produit, sauf si celle-ci ressort de la présentation du produit, la liste des ingrédients conforme à la nomenclature commune arrêtée par la Commission européenne ainsi que les règles particulières applicables à la publicité pour ces produits lorsqu’il est fait référence à l’expérimentation animale ; 2° Les modalités de présentation et le contenu de la déclaration prévue au premier alinéa de l’article L. 5131-2 ; 3° Le contenu du dossier mentionné à l’article L. 5131-6 et les conditions de protection du secret des informations figurant dans ce dossier notamment celles relatives à des composants ou ingrédients délivrés par des fournisseurs exclusifs et responsables ; 4° Les règles relatives à la composition des produits cosmétiques ; 5° Les conditions de transmission aux centres antipoison et de protection du secret des informations mentionnées à l’article L. 5131-7 ; 6° Les modalités d’application du I de l’article L. 5131-9 ; 7° Les modalités d’application de l’article L. 5131-10 en ce qui concerne le contenu des informations demandées, les règles assurant le respect de leur confidentialité et le délai maximum de réponse ; 8° Les conditions de mise à la disposition du public des informations mentionnées à l’article L. 5131-7-1. Des décrets fixent les conditions d’utilisation professionnelle des produits cosmétiques lorsque cette utilisation est susceptible de comporter des dangers ou des inconvénients. |
Abrogé |
|
II. – Le chapitre Ier du titre III du livre IV de la cinquième partie du code de la santé publique est ainsi modifié : |
||
1° L’article L. 5431-2 est rem-placé par les dispositions suivantes : |
||
Art. L. 5431-2. – Est puni de deux ans d’emprisonnement et de 30000 euros d’amende, le fait : |
« Art. L. 5431-2. – Est puni de deux ans d’emprisonnement et de 30 000 € d’amende, le fait : |
|
1° D’ouvrir ou d’exploiter un établissement de fabrication, de conditionnement ou d’importation de produits cosmétiques, à l’exception de ceux mentionnés à l’article L. 5131-3, ou d’étendre l’activité d’un établissement à de telles opérations, sans qu’ait été faite au préalable la déclaration à l’Agence nationale de sécurité du médicament et des produits de santé ou sans qu’aient été déclarées les modifications des éléments figurant dans la déclaration initiale ; |
« 1° Pour toute personne qui fabrique ou conditionne des produits cosmétiques, d’ouvrir ou d’exploiter un établissement de fabrication ou de conditionnement de ces produits, ou d’étendre l’activité d’un établissement à de telles opérations, sans qu’ait été faite au préalable la déclaration prévue à l’article L. 5131-2 à l’Agence nationale de sécurité du médicament et des produits de santé ou sans qu’aient été déclarées les modifications des éléments figurant dans la déclaration ; |
« 1° Pour … … été communiquées les modifications des éléments constitutifs de la déclaration. Amendement AS27 |
2° De diriger un établissement mentionné au 1° ci-dessus sans avoir désigné la ou les personnes qualifiées responsables conformément à l’article L. 5131-2 ; |
« 2° Pour la personne responsable de la mise sur le marché d’un produit cosmétique, telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009, de ne pas respecter les obligations de notification à la Commission européenne en ne transmettant pas les informations mentionnées aux paragraphes 1 et 2 de l’article 13 et au paragraphe 3 de l’article 16 de ce même règlement ; |
|
3° Pour le responsable de la mise sur le marché national d’un produit cosmétique, de ne pas transmettre aux centres antipoison les informations prévues à l’article L. 5131-7 ; 4° De mettre sur le marché des produits cosmétiques ou de réaliser des expérimentations animales portant sur des produits cosmétiques finis ou sur des ingrédients ou des combinaisons d’ingrédients en méconnaissance des interdictions prévues à l’article L. 5131-7-2. |
« 3° Pour la personne responsable de la mise sur le marché d’un produit cosmétique, telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009, de mettre sur le marché des produits cosmétiques ou de réaliser des expérimentations animales portant sur des produits cosmétiques finis ou sur des ingrédients ou des combinaisons d’ingrédients en méconnaissance des interdictions prévues au paragraphe 1 de l’article 18 de ce règlement. » ; |
|
Art. L. 5431-3. – Les personnes physiques coupables des infractions définies à l’article L. 5431-2 encourent également les peines complémentaires suivantes : 1° La diffusion de la décision de condamnation et celle d’un ou plusieurs messages informant le public de cette décision, dans les conditions prévues à l’article 131-35 du code pénal ; 2° L’affichage de la décision prononcée, dans les conditions et sous les peines prévues à l’article 131-35 du même code ; 3° La confiscation de la chose qui a servi ou était destinée à commettre l’infraction ou du produit de la vente de cette chose, dans les conditions prévues à l’article 131-21 du même code ; 4° La fermeture définitive ou pour une durée de cinq ans au plus des établissements de l’entreprise ayant servi à commettre les faits incriminés, dans les conditions prévues à l’article 131-33 du même code ; |
||
5° L’interdiction de fabriquer, de conditionner, d’importer, de mettre sur le marché des produits cosmétiques pour une durée maximum de cinq ans. |
1° bis Au dernier alinéa de l’article L. 5431-3, le mot : « importer, » est remplacé par les mots : « importer et ». Amendement AS31 | |
2° L’article L. 5431-5 est ainsi modifié : |
||
Art. L. 5431-5. – Est puni de deux ans d’emprisonnement et de 30 000 euros d’amende le fait pour les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un Etat qui n’est ni membre de l’Union européenne, ni partie à l’accord sur l’Espace économique européen de mettre sur le marché à titre gratuit ou onéreux un produit cosmétique qui n’est pas conforme aux règles relatives aux substances entrant dans la composition de ce produit, telles qu’elles résultent du 4° de l’article L. 5131-11. |
a) Les mots : « les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un État qui n’est ni membre de l’Union européenne, ni partie à l’accord sur l’Espace économique européen » sont remplacés par les mots : « la personne responsable telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009 » ; b) Les mots : « telles qu’elles résultent du 4° de l’article L. 5131-11 » sont remplacés par les mots : « prévues à l’article 14 et aux paragraphes 1 et 2 de l’article 15 du règlement (CE) n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques » ; |
|
3° L’article L. 5431-6 est ainsi modifié : |
||
Art. L. 5431-6. – Est puni d’un an d’emprisonnement et de 15 000 euros d’amende le fait pour les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un Etat qui n’est ni membre de l’Union européenne, ni partie à l’accord sur l’Espace économique européen : |
a) Au premier alinéa, les mots : « les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un État qui n’est ni membre de l’Union, ni partie à l’accord sur l’Espace économique européen » sont remplacés par les mots : « la personne responsable telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009 » ; |
|
1° De mettre sur le marché à titre gratuit ou onéreux un produit cosmétique sans tenir à disposition des autorités de contrôle le dossier mentionné au troisième alinéa de l’article L. 5131-6 ; |
b) Au 1°, les mots : « de contrôle le dossier mentionné au troisième alinéa de l’article L. 5131-6 » sont remplacés par les mots : « de contrôle mentionnées à l’article L. 5431-1, à l’adresse indiquée sur l’étiquetage de ce produit, le dossier prévu au paragraphe 1 de l’article 11 du règlement (CE) n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques » ; |
b) Après le mot : « contrôle », la fin du 1° est ainsi rédigée : « mentionnées à l’article L. 5431-1, à l’adresse indiquée sur l’étiquetage de ce produit, le dossier d’information prévu au paragraphe 1 de l’article 11 du même règlement ; » Amendement AS14 |
2° De mettre sur le marché à titre gratuit ou onéreux un produit cosmétique sans que le dossier mentionné au troisième alinéa de l’article L. 5131-6 comporte les mentions obligatoires prévues par le 3° de l’article L. 5131-11 ; |
c) Au 2°, les mots : « mentionné au troisième alinéa de l’article L. 5131-6 » et les mots : « par le 3° de l’article L. 5131-11 » sont respectivement remplacés par les mots : « mentionné à l’article 11 de ce règlement » et les mots : « au paragraphe 2 de cet article » ; |
c) Le 2° est ainsi modifié : – après le mot : « dossier », sont insérés les mots : « d’information » ; – la référence : « au troisième alinéa de l’article L. 5131-6 » est remplacée par la référence : « à l’article 11 dudit règlement » ; – à la fin, la référence : « par le 3° de l’article L. 5131-11 » est remplacée par la référence : « au paragraphe 2 du même article » ; Amendement AS14 |
3° De ne pas transmettre à l’Agence nationale de sécurité du médicament et des produits de santé , sur sa demande, l’une des informations mentionnées à l’article L. 5131-10. |
d) Le 3° est abrogé ; |
|
4° L’article L. 5431-7 est ainsi modifié : |
||
Art. L. 5431-7. – Le fait, pour les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un Etat qui n’est ni membre de l’Union européenne, ni partie à l’accord sur l’Espace économique européen, de mettre sur le marché à titre gratuit ou onéreux un produit cosmétique dont le récipient ou l’emballage ne comporte pas l’une des mentions prévues au deuxième alinéa de l’article L. 5131-6 et au 1° de l’article L. 5131-11 est puni de 15 000 euros d’amende. |
a) Les mots : « les fabricants, leurs représentants, les personnes pour le compte desquelles les produits cosmétiques sont fabriqués ou les responsables de la mise sur le marché des produits cosmétiques importés d’un État qui n’est ni membre de l’Union européenne, ni partie à l’accord sur l’Espace économique européen » sont remplacés par les mots : « la personne responsable telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009 » ; b) Les mots : « au deuxième alinéa de l’article L. 5131-6 et au 1° de l’article L. 5131-11 » sont remplacés par les mots : « à l’article 19 du règlement (CE) n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques » ; |
|
5° Après l’article L. 5431-7, sont ajoutés deux articles ainsi rédigés : |
||
« Art. L. 5431-8. – Le fait, pour la personne responsable telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009 et les distributeurs tels que définis par l’article 2 de ce même règlement, de ne pas signaler à l’Agence nationale de sécurité du médicament et des produits de santé, dès qu’ils en ont connaissance et par tout moyen, tous les effets indésirables graves au sens du point p du paragraphe 1 de l’article 2 de ce règlement dans les conditions mentionnées au paragraphe 1 de son article 23, est puni de deux ans d’emprisonnement et de 30 000 € d’amende. |
« Art. L. 5431-8. – … … définis au e du paragraphe 1 de l’article 2 de ce même règlement, de ne pas signaler à l’Agence nationale de sécurité du médicament et des produits de santé, sans délai et par tout … … p du même paragraphe 1, dans les conditions … …d’amende. Amendements AS15 et AS3 | |
« Est puni des mêmes peines le fait, pour le professionnel de santé ayant eu connaissance, dans l’exercice de ses fonctions, d’un effet indésirable grave au sens du point p du paragraphe 1 de l’article 2 du règlement (CE) n° 1223/2009 du 30 novembre 2009, de s’abstenir de le signaler sans délai à l’agence. |
||
« Art. L. 5431-9. – Le fait, pour la personne responsable telle que déterminée par les troisième à sixième alinéas de l’article 4 du règlement (CE) n° 1223/2009 du 30 novembre 2009, de ne pas transmettre à l’Agence nationale de sécurité du médicament et des produits de santé, malgré la mise en demeure effectuée dans les conditions fixées par l’article L. 5131-6, l’une des informations mentionnées à l’arti-cle 24 du règlement (CE) n° 1223/2009 du Parlement européen et du Conseil du 30 novembre 2009 relatif aux produits cosmétiques est puni d’un an d’emprisonnement et 15 000 € d’amende. » |
||
III. – Le chapitre X du titre III du livre Ier de la cinquième partie du code de la santé publique est ainsi modifié : |
||
1° L’article L. 513-10-2 est remplacé par les dispositions suivantes : |
||
Art. L. 513-10-2. – Les disposi-tions prévues pour les produits cosmétiques aux premier, troisième et quatrième alinéas de l’article L. 5131-2 et aux articles L. 5131-4 et L. 5131-6 à L. 5131-10 sont applicables aux produits de tatouage. |
« Art. L. 513-10-2. – L’ouverture et l’exploitation de tout établissement de fabrication, de conditionnement ou d’importation, même à titre accessoire, de produits de tatouage, de même que l’extension de l’activité d’un établissement à de telles opérations, sont subordonnées à une déclaration auprès de l’Agence nationale de sécurité du médicament et des produits de santé. |
|
La déclaration prévue au premier alinéa de l’article L. 5131-2 est effectuée par le fabricant, ou par son représentant ou par la personne pour le compte de laquelle les produits de tatouage sont fabriqués, ou par le responsable de la mise sur le marché des produits de tatouage importés. Elle indique les personnes qualifiées responsables désignées en application du quatrième alinéa de l’article L. 5131-2. |
« Cette déclaration est faite par la personne responsable de la mise sur le marché des produits de tatouage, qui est, selon le cas, le fabricant ou son représentant, la personne pour le compte de laquelle les produits de tatouage sont fabriqués ou la personne qui met sur le marché les produits de tatouage importés. |
|
« Toute modification des éléments constitutifs de la déclaration est communiquée à l’agence. |
||
« La personne responsable de la mise sur le marché des produits de tatouage désigne une ou plusieurs personnes qualifiées responsables de la fabrication, du conditionnement, de l’importation, des contrôles de qualité, de l’évaluation de la sécurité pour la santé humaine, de la détention et de la surveillance des stocks de matières premières et de produits finis. Ces personnes doivent posséder des connaissances scientifiques suffisantes attestées par des diplômes, titres ou certificats figurant sur une liste établie par arrêté des ministres chargés de l’artisanat, de l’enseignement supérieur, de l’industrie et de la santé ou justifier d’une expérience pratique appropriée dont la durée et le contenu sont déterminés dans les mêmes conditions. » ; |
||
2° Les deuxième et troisième phrases de l’article L. 513-10-3 sont remplacées par les dispositions suivantes : |
||
Art. L. 513-10-3. – La fabrication des produits de tatouage doit être réalisée en conformité avec les bonnes pratiques de fabrication dont les principes sont définis par arrêté des ministres chargés de la consommation et de la santé, pris sur proposition de l’Agence nationale de sécurité du médicament et des produits de santé. L’évaluation de la sécurité pour la santé humaine de ces produits doit être exécutée en conformité avec les bonnes pratiques de laboratoire dont les principes sont définis dans les mêmes conditions. Les règles générales relatives aux modalités d’inspection et de vérification des bonnes pratiques de laboratoire ainsi qu’à la délivrance de documents attestant de leur respect sont définies par arrêté des ministres chargés de la consommation et de la santé, pris sur proposition de l’Agence nationale de sécurité du médicament et des produits de santé. |
« L’évaluation de la sécurité pour la santé humaine de ces produits doit être exécutée en conformité avec les bonnes pratiques de laboratoire dont les principes sont publiés par l’Agence nationale de sécurité du médicament et des produits de santé, de même que les règles applicables à l’inspection et à la vérification des bonnes pratiques de laboratoire. Un arrêté des ministres chargés de la consommation et de la santé, pris sur proposition de l’agence, définit les règles relatives à la délivrance de documents attestant du respect de ces bonnes pratiques. » ; |
« L’évaluation de la sécurité pour la santé humaine de ces produits est exécutée … … pratiques. » ; Amendement AS16 |
3° L’article L. 513-10-4 est remplacé par les dispositions suivantes : |
||
Art. L. 513-10-4. – Les modalités d’application du présent chapitre et les règles relatives à la composition ainsi que les exigences de qualité et de sécurité des produits de tatouage sont déterminées par décret en Conseil d’Etat. |
« Art. L. 513-10-4. – Les pro-duits de tatouage mis sur le marché ne doivent pas nuire à la santé humaine lorsqu’ils sont appliqués dans les conditions normales ou raisonnablement prévisibles d’utilisation compte tenu, notamment, de la présentation du produit, des mentions portées sur l’étiquetage ainsi que de toute autre information destinée aux consommateurs. » ; |
|
4° Après l’article L. 513-10-4, il est inséré six articles ainsi rédigés : |
||
« Art. L. 513-10-5. – Un produit de tatouage ne peut être mis sur le marché à titre gratuit ou onéreux que : |
« Art. L. 513-10-5. – … … que s’il remplit les conditions suivantes : | |
« 1° Si son récipient et son emballage comportent le nom ou la raison sociale et l’adresse de la personne responsable de la mise sur le marché du produit, ainsi que les autres mentions prévues par voie réglementaire ; |
« 1° Son récipient … … réglementaire ; | |
« 2° Et si la personne responsable de la mise sur le marché du produit tient effectivement à la disposition des autorités de contrôle, à l’adresse mentionnée ci-dessus, un dossier rassemblant toutes les informations utiles au regard des dispositions des articles L. 513-10-3 et L. 513-10-4, sur la formule qualitative et quantitative, les spécifications physico-chimiques et microbiologiques, les conditions de fabrication et de contrôle, l’évaluation de la sécurité pour la santé humaine et les effets indésirables de ce produit. |
« 2° La personne … … produit. Amendement AS17 | |
« Art. L. 513-10-6. – La mise sur le marché à titre gratuit ou onéreux d’un produit de tatouage est subordonnée à la transmission aux centres antipoison mentionnés à l’article L. 6141-4, désignés par arrêté des ministres chargés de la consommation, de l’industrie et de la santé, d’informations adéquates et suffisantes concernant les substances utilisées dans ce produit. |
« Art. L. 513-10-6. – … … arrêté conjoint des ministres … … produit. | |
« La liste de ces informations est fixée par arrêté des ministres chargés de la consommation, de l’industrie et de la santé. |
« La … arrêté conjoint des ministres … … santé. Amendement AS18 | |
« Art. L. 513-10-7. – La personne responsable de sa mise sur le marché met à la disposition du public, par des moyens appropriés, y compris des moyens électroniques, les informations liées à la composition et aux effets indésirables du produit de tatouage définies par voie réglementaire. |
« Art. L. 513-10-7. – La personne responsable de la mise sur le marché du produit de tatouage met … … liées à sa composition et aux effets indésirables de ce produit, définies par voie réglementaire. Amendement AS19 | |
« Art. L. 513-10-8. – I. – La personne responsable de la mise sur le marché d’un produit de tatouage est tenue de participer au système national de vigilance exercé sur les produits de tatouage en déclarant sans délai à l’Agence nationale de sécurité du médicament et des produits de santé les effets indésirables graves susceptibles de résulter de l’utilisation d’un produit de tatouage dont elle a connaissance. Elle déclare, en outre, les autres effets indésirables dont elle a connaissance. Est un effet indésirable grave une réaction nocive et non prévisible, qu’elle se produise dans les conditions normales d’emploi du produit chez l’homme ou qu’elle résulte d’un mésusage, qui est de nature à justifier une hospitalisation ou entraîne une incapacité fonctionnelle temporaire ou permanente, une invalidité, une mise en jeu du pronostic vital immédiat, un décès ou une anomalie ou une malformation congénitale. |
« Art. L. 513-10-8. – … … connaissance. Elle lui déclare … … congénitale. Amendement AS20 | |
« Cette obligation est réputée remplie par le respect de l’obligation d’information prévue par les dispositions de l’article L. 221-1-3 du code de la consommation. Dans ce cas, l’Agence nationale de sécurité du médicament et des produits de santé est informée sans délai par les autorités administratives compétentes men-tionnées au même article. |
L’obligation mentionnée au premier alinéa du présent I est réputée … … article. Amendement AS21 | |
« II. – Tout professionnel de santé ayant connaissance d’un effet indésirable grave susceptible de résulter de l’utilisation d’un produit de tatouage le déclare sans délai à l’Agence nationale de sécurité du médicament et des produits de santé. Il déclare, en outre, les autres effets indésirables dont il a connaissance. Dans sa déclaration, il précise si l’effet indésirable résulte d’un mésusage. |
« II. – Tout … … grave, au sens du I du présent article, suceptible … … santé. Il lui déclare … … mésusage et décrit les conditions dans lesquelles le tatouage a été pratiqué. Amendements AS22, AS30 et AS4 | |
« Toute personne qui réalise des tatouages à titre professionnel ayant connaissance d’un effet indésirable grave susceptible de résulter de l’utilisation d’un produit de tatouage le déclare sans délai à l’Agence nationale de sécurité du médicament et des produits de santé. Il déclare, en outre, les autres effets indésirables dont il a connaissance. Dans sa déclaration, il précise si l’effet indésirable résulte d’un mésusage. |
« Toute … … grave, au sens du I du présent article, susceptible … … santé. Il lui déclare … … mésusage et décrit les conditions dans lesquelles le tatouage a été pratiqué. Amendements AS22, AS30 et AS4 | |
« Tout consommateur peut décla-rer des effets indésirables à l’Agence nationale de sécurité du médicament et des produits de santé, en faisant état, le cas échéant, d’un mésusage. |
« Tout … … mésusage et en décrivant les conditions dans lesquelles le tatouage a été pratiqué. Amendement AS4 | |
« Art. L. 513-10-9. – La personne responsable de la mise sur le marché d’un produit de tatouage est tenue, en cas de doute sérieux sur l’innocuité d’une ou de plusieurs substances, de fournir au directeur général de l’Agence nationale de sécurité du médicament et des produits de santé, lorsqu’il lui en fait la demande motivée, la liste de ses produits de tatouage dans la composition desquels entrent une ou plusieurs substances désignées par lui ainsi que la quantité de chacune de ces substances présentes dans le produit. |
||
« L’Agence prend toute mesure pour protéger la confidentialité des informations qui lui sont transmises au titre du présent article. |
||
« Art. L. 513-10-10. – Un décret en Conseil d’État détermine les modalités d’application du présent chapitre, notamment : |
||
« 1° Les modalités de présentation et le contenu de la déclaration prévue à l’article L. 513-10-2 ; |
||
« 2° Les mentions que doivent comporter le récipient et l’emballage d’un produit de tatouage en application du deuxième alinéa de l’article L. 513-10-5 ; |
||
« 3° Le contenu et les modalités de conservation du dossier mentionné au troisième alinéa de l’article L. 513-10-5 ; |
||
« 4° Les informations que la personne responsable de la mise sur le marché met à la disposition du public en application de l’article L. 513-10-7 ; |
||
« 5° Les modalités de mise en œuvre du système national de vigilance exercé sur les produits de tatouage prévu à l’article L. 513-10-8 ; |
||
« 6° Les exigences de qualité et de sécurité des produits de tatouage et les règles relatives à leur composition. » |
||
IV. – Le chapitre VII du titre III du livre IV de la cinquième partie du code de la santé publique est ainsi modifié : |
||
1° L’article L. 5437-2 est remplacé par les dispositions suivantes : |
||
Art. L. 5437-2. – Les infractions prévues à l’article L. 5431-2 sont applicables aux produits de tatouage et sont punies des peines prévues, pour les personnes physiques et morales, aux articles L. 5431-2 à L. 5431-4. |
« Art. L. 5437-2. – Est puni de deux ans d’emprisonnement et de 30 000 € d’amende le fait : |
|
« 1° Pour la personne responsable d’établissement de fabrication, de conditionnement ou d’importation de produits de tatouage, d’ouvrir, exploiter ou étendre l’activité d’un établissement à de telles opérations, sans qu’ait été faite au préalable la déclaration à l’Agence nationale de sécurité du médicament et des produits de santé, ou sans qu’aient été déclarées les modifications des éléments figurant dans la déclaration, telles que prévues aux deuxième et troisième alinéas de l’article L. 513-10-2 ; |
||
« 2° Pour la personne responsable d’établissement de fabrication, de conditionnement ou d’importation de produits de tatouage, de diriger un établissement mentionné au 1° sans avoir désigné la ou les personnes qualifiées responsables conformément à l’article L. 513-10-2 ; |
||
« 3° Pour la personne responsable de la mise sur le marché national du produit de tatouage au sens de l’article L. 513-10-2, de ne pas transmettre aux centres antipoison les informations mentionnées à l’article L. 513-10-5. » ; |
« 3° Pour … … L. 513-10-6. » Amendement AS23 | |
2° Après l’article L. 5437-2, il est inséré trois articles ainsi rédigés : |
||
« Art. L. 5437-3. – Les personnes physiques coupables des infractions définies à l’article L. 5437-2 encourent également les peines complémentaires suivantes : |
||
« 1° La diffusion de la décision de condamnation et celle d’un ou plusieurs messages informant le public de cette décision, dans les conditions prévues à l’article 131-35 du code pénal ; |
||
« 2° L’affichage de la décision prononcée, dans les conditions et sous les peines prévues à l’article 131-35 du même code ; |
||
« 3° La confiscation de la chose qui a servi ou était destinée à commettre l’infraction ou du produit de la vente de cette chose, dans les conditions prévues à l’article 131-21 du même code ; |
||
« 4° La fermeture définitive ou pour une durée de cinq ans au plus des établissements de l’entreprise ayant servi à commettre les faits incriminés, dans les conditions prévues à l’article 131-33 du même code ; |
||
« 5° L’interdiction de fabriquer, de conditionner, d’importer, de mettre sur le marché des produits de tatouage pour une durée maximum de cinq ans. |
« 5° L’interdiction … … d’importer et de mettre … … cinq ans. Amendement AS28 | |
« Art. L. 5437-4. – Les personnes morales déclarées responsables pénalement, dans les conditions prévues par l’article 121-2 du code pénal, des infractions définies à l’article L. 5437-2 encourent, outre l’amende suivant les modalités prévues par l’article 131-38 du code pénal : |
||
« 1° La confiscation de la chose qui a servi ou était destinée à commettre l’infraction ou de la chose qui en est le produit, dans les conditions prévues au 8° de l’article 131-39 du même code ; |
||
« 2° L’affichage de la décision prononcée ou la diffusion de celle-ci soit par la presse écrite, soit par tout moyen de communication audiovisuelle, dans les conditions prévues au 9° de l’article 131-39 du même code ; |
« 2° L’affichage … … communication électronique, dans … … code ; Amendement AS24 | |
« 3° La fermeture définitive ou pour une durée de cinq ans au plus des établissements de l’entreprise ayant servi à commettre les faits incriminés, dans les conditions prévues au 4° de l’article 131-39 du même code. |
||
« Art. L. 5437-5. – Le fait, pour la personne responsable de la mise sur le marché du produit de tatouage au sens de l’article L. 513-10-2, de ne pas signaler, dès qu’elle en a connaissance et par tout moyen à l’Agence nationale de sécurité du médicament et des produits de santé les effets indésirables graves dans les conditions mentionnées à l’article L. 513-10-8 est puni de deux ans d’emprisonnement et de 30 000 € d’amende. |
« Art. L. 5437-5. – … … mentionnées au I de l’article … … d’amende. Amendement AS25 | |
« Est puni des mêmes peines le fait, pour le professionnel de santé ou la personne qui réalise des tatouages à titre professionnel ayant eu personnellement connaissance, dans l’exercice de ses fonctions, d’un effet indésirable grave, de s’abstenir de le signaler sans délai à l’agence. » |
« Est … … grave, au sens du I de l’article L. 513-10-8, de s’abstenir … … l’agence. » Amendement AS25 | |
Art. L. 5122-14. – La publicité pour les produits autres que les médicaments présentés comme favorisant le diagnostic, la prévention ou le traitement des maladies, des affections relevant de la pathologie chirurgicale et des dérèglements physiologiques, le diagnostic ou la modification de l’état physique ou physiologique, la restauration, la correction ou la modification des fonctions organiques est soumise aux dispositions du premier alinéa de l’article L. 5122-2 et des articles L. 5122-8 et L. 5122-9. |
V. – L’article L. 5122-14 du code de la santé publique est abrogé. |
|
VI. – L’article L. 5131-7 du même code, dans sa rédaction résultant de la présente loi, est abrogé à compter du 12 juillet 2020. |
||
Article 4 |
Article 4 | |
I. – Après l’article L. 4362-9 du code de la santé publique, il est inséré un article L. 4362-9-1 ainsi rédigé : |
(Sans modification) | |
« Art. L. 4362-9-1. – I. – Les conditions de première délivrance de lentilles correctrices sont déterminées par décret en Conseil d’État. |
||
« II. – Lors de la vente en ligne de lentilles correctrices, les prestataires concernés permettent au patient d’obtenir informations et conseils auprès d’un professionnel de santé qualifié. Un décret en Conseil d’État détermine les modalités d’application du présent alinéa et fixe les mentions et informations devant figurer sur le site internet. » |
||
II. – L’article L. 4363-4 du même code est complété par un alinéa ainsi rédigé : |
||
Art. L. 4363-4. – Est puni de 3750 euros d’amende le fait : 1° De diriger ou de gérer, sans remplir les conditions requises pour l’exercice de la profession d’opticien-lunetier, un établissement commercial dont l’objet principal est l’optique-lunetterie, une succursale d’un tel établissement ou un rayon d’optique-lunetterie des magasins ; 2° De colporter des verres correcteurs d’amétropie ; 3° De délivrer un verre correcteur à une personne âgée de moins de 16 ans sans ordonnance médicale. |
||
« 4° De délivrer ou de vendre des lentilles correctrices en méconnaissance des dispositions relatives aux conditions de première délivrance et aux obligations à la charge des prestataires de vente en ligne mentionnées à l’article L. 4362-9-1. » |
||
Ordonnance n° 2012-1427 du 19 décembre 2012 |
Article 5 |
Article 5 |
Cf. Annexe |
I. – L’ordonnance n° 2012-1427 du 19 décembre 2012 relative au renforcement de la sécurité de la chaîne d’approvisionnement des médicaments, à l’encadrement de la vente de médicaments sur internet et à la lutte contre la falsification de médicaments est ratifiée. |
(Sans modification) |
Code de la santé publique |
II. – Le code de la santé publique est ainsi modifié : |
|
Art. L. 5124-1. – La fabrication, l’importation, l’exportation et la distribution en gros de médicaments, produits et objets mentionnés à l’article L. 4211-1, la fabrication, l’importation et la distribution des médicaments expérimentaux, à l’exception des préparations de thérapie génique et des préparations de thérapie cellulaire xénogénique, ainsi que l’exploitation de spécialités pharmaceutiques ou autres médicaments, de générateurs, trousses ou précurseurs définis aux 8°, 9° et 10° de l’article L. 5121-1 ne peuvent être effectuées que dans des établissements pharmaceutiques régis par le présent chapitre. |
||
Les personnes se livrant à une activité de courtage de médicaments mentionnée à l’article L. 5121-19 ne sont pas soumises aux dispositions du présent chapitre. |
1° À l’article L. 5124-1, la référence à l’article L. 5121-19 est remplacée par la référence à l’article L. 5124-19 ; |
|
Art. L. 5125-33. – On entend par commerce électronique de médicaments l’activité économique par laquelle le pharmacien propose ou assure à distance et par voie électronique la vente au détail et la dispensation au public des médicaments à usage humain et, à cet effet, fournit des informations de santé en ligne. |
||
L’activité de commerce élec-tronique est réalisée à partir du site internet d’une officine de pharmacie. |
||
La création et l’exploitation d’un tel site sont exclusivement réservées aux pharmaciens suivants : |
||
1°Pharmacien titulaire d’une officine ; |
||
2° Pharmacien gérant d’une pharmacie mutualiste ou de secours minière, exclusivement pour leurs membres. |
||
Le pharmacien titulaire de l’officine ou gérant d’une pharmacie mutualiste ou de secours minière est responsable du contenu du site internet qu’il édite et des conditions dans lesquelles l’activité de commerce électronique de médicaments s’exerce. |
||
Les pharmaciens adjoints ayant reçu délégation du pharmacien d’officine peuvent participer à l’exploitation du site internet de l’officine de pharmacie. |
2° Au septième alinéa de l’article L. 5125-33, les mots : « du pharmacien d’officine » sont remplacés par les mots : « de l’un des pharmaciens mentionnés à l’alinéa précédent » ; |
|
Les pharmaciens remplaçant de titulaires d’officine ou gérants d’officine après décès du titulaire peuvent exploiter le site internet de l’officine créé antérieurement par le titulaire de l’officine. |
||
3° L’article L. 5125-34 est remplacé par les dispositions suivantes : |
||
Art. L. 5125-34. – Seuls peuvent faire l’objet de l’activité de commerce électronique les médicaments de médication officinale qui peuvent être présentés en accès direct au public en officine, ayant obtenu l’autorisation de mise sur le marché mentionnée à l’article L. 5121-8 ou un des enregistrements mentionnés aux articles L. 5121-13 et L. 5121-14-1. |
« Art. L. 5125-34. – Seuls peuvent faire l’objet de l’activité de commerce électronique les médicaments qui ne sont pas soumis à prescription obligatoire. » ; |
|
Art. L. 5125-39. – En cas de manquement aux règles applicables au commerce électronique prévues par les dispositions du présent chapitre et aux bonnes pratiques de dispensation mentionnées à l’article L. 5121-5 par l’un des pharmaciens mentionnés à l’article L. 5125-33, le directeur général de l’agence régionale de santé territorialement compétente peut après, sauf en cas d’urgence, avoir mis en demeure, dans un délai qu’il fixe et qui ne peut être inférieur à huit jours, l’auteur du manquement de se conformer à ses prescriptions et de présenter ses observations : |
||
1° Prononcer la fermeture tempo-raire du site internet de commerce électronique de médicaments pour une durée maximale de cinq mois ; |
||
2° Prononcer une amende administrative à l’encontre de l’auteur du manquement et, le cas échéant, assortir cette amende d’une astreinte journalière qui ne peut être supérieure à 1 000 € par jour lorsque l’auteur de l’infraction ne s’est pas conformé à ses prescriptions à l’issue d’un délai fixé par une mise en demeure. Le montant de l’amende administrative ne peut être supérieur à 30 % du chiffre d’affaires réalisé par la pharmacie dans le cadre de l’activité de commerce électronique, dans la limite d’un million d’euros. |
4° Au 2° de l’article L. 5125-39, après le mot : « réalisé », sont insérés les mots : « lors du dernier exercice clos » ; |
|
Lorsqu’au terme de la durée de fermeture du site internet le pharmacien ne s’est pas mis en conformité avec les règles applicables, le directeur général de l’agence régionale de santé peut prononcer dans les mêmes conditions une nouvelle fermeture. |
||
L’agence régionale de santé informe le conseil de l’ordre compétent de la mise en œuvre de la procédure prévue au présent article. |
||
Art. L. 5438-2. – L’Agence na-tionale de sécurité du médicament et des produits de santé peut prononcer une amende administrative à l’encontre de l’auteur d’un manquement mentionné à l’article L. 5438-1. Elle peut assortir cette amende d’une astreinte journalière qui ne peut être supérieure à 2 500 € par jour lorsque l’auteur du manquement ne s’est pas conformé à ses prescriptions à l’issue du délai fixé par une mise en demeure. |
||
Le montant de l’amende prononcée pour les manquements mentionnés à l’article L. 5438-1 ne peut être supérieur à 10 % du chiffre d’affaires réalisé, dans la limite d’un million d’euros. |
5° Au dernier alinéa de l’article L. 5438-2, après le mot : « réalisé », sont insérés les mots : « lors du dernier exercice clos, » ; |
|
Art. L. 5438-6. – La tentative des délits prévus à l’article L. 5438-2 est punie des mêmes peines. |
6° À l’article L. 5438-6, la référence à l’article L. 5438-2 est remplacée par la référence à l’article L. 5438-4 ; |
|
7° L’article L. 5438-7 est remplacé par les dispositions suivantes : |
||
Art. L. 5438-7. – Les personnes morales déclarées pénalement respon-sables, dans les conditions prévues à l’article 121-2 du code pénal, des infractions prévues au présent chapitre encourent, outre l’amende suivant les modalités prévues à l’article 131-38 du même code, les peines prévues aux 2° à 9° de l’article 13139 de ce code. |
« Art. L. 5438-7. – Pour les infractions pénales mentionnées au présent chapitre, les personnes physiques encourent également les peines complémentaires suivantes : |
|
« 1° L’affichage ou la diffusion de la décision prononcée, dans les conditions et sous les peines prévues à l’article 131-35 du code pénal ; |
||
« 2° L’interdiction temporaire ou définitive d’exercer une ou plusieurs professions régies par le présent code ou toute autre activité professionnelle ou sociale à l’occasion de l’exercice de laquelle l’infraction a été commise, suivant les modalités prévues à l’article 131-27 du même code ; |
||
« 3° La confiscation de la chose qui a servi ou était destinée à commettre l’infraction ou de la chose qui en est le produit, en application de l’article 131-21 du même code. » ; |
||
8° Après l’article L. 5438-7, il est ajouté un article L. 5438-8 ainsi rédigé : |
||
« Art. L. 5438-8. – Les personnes morales déclarées pénalement respon-sables, dans les conditions prévues à l’article 121-2 du code pénal, des infractions prévues au présent chapitre encourent, outre l’amende suivant les modalités prévues à l’article 131-38 du même code, les peines prévues aux 2° à 9° de l’article 131-39 de ce code. » |
||
Article 6 |
Article 6 | |
I. – L’article L. 5121-9-4 du code de la santé publique est remplacé par les dispositions suivantes : |
||
Art. L. 5121-9-4. – Le titulaire de l’autorisation de mise sur le marché qui arrête la commercialisation d’un médicament dans un autre État que la France alors que ce produit reste commercialisé en France doit en informer immédiatement l’Agence nationale de sécurité du médicament et des produits de santé et lui communiquer le motif de cet arrêt de commercialisation. |
« Art. L. 5121-9-4. – Le titulaire de l’autorisation de mise sur le marché d’un médicament informe, immédia-tement et de manière motivée, l’Agence nationale de sécurité du médicament et des produits de santé de toute action engagée, en France ou dans un autre État membre, pour suspendre ou arrêter la commercialisation de ce médicament, pour solliciter le retrait de cette autorisation ou pour ne pas en demander le renouvellement, en précisant notamment si son action est fondée sur l’un des motifs mentionnés aux 1° à 5° de l’article L. 5121-9. Si son action est fondée sur l’un des motifs précités, il en informe également l’Agence européenne des médicaments. |
« Art. L. 5121-9-4. – … … membre de l’Union européenne, pour … … médicaments. Amendement AS26 |
« Lorsque l’une des actions mentionnées à l’alinéa précédent est engagée dans un pays tiers et qu’elle est fondée sur l’un des motifs mentionnés aux 1° à 5° de l’article L. 5121-9, le titulaire de l’autorisation de mise sur le marché en informe de manière motivée l’Agence nationale de sécurité du médicament et des produits de santé et l’Agence européenne des médicaments. » |
||
II. – Le premier alinéa de l’article L. 5124-6 du code de la santé publique est ainsi modifié : |
||
Art. L. 5124-6. – L’entreprise pharmaceutique exploitant un médicament ou produit soumis aux dispositions du chapitre Ier du présent titre qui prend la décision d’en suspendre ou d’en cesser la commer-cialisation ou qui a connaissance de faits susceptibles d’entraîner la suspension ou la cessation de cette commercialisation en informe au moins un an avant la date envisagée ou prévisible l’Agence nationale de sécurité du médicament et des produits de santé si ce médicament est utilisé dans une ou des pathologies graves dans lesquelles elle ne disposerait pas d’alternatives disponibles sur le marché français. La cessation de commer-cialisation ne peut intervenir avant la fin du délai nécessaire pour mettre en place les solutions alternatives permettant de couvrir ce besoin. Si le médicament n’est pas utilisé dans une ou des pathologies graves dans lesquelles elle ne disposerait pas d’alternatives disponibles sur le marché français, la notification doit avoir lieu au plus tard deux mois avant la suspension ou l’arrêt de commercialisation. En cas d’urgence nécessitant que la suspension ou l’arrêt intervienne avant le terme des délais fixés ci-dessus, l’entreprise en informe immédiatement l’agence en justifiant de cette urgence. Elle doit en outre informer l’Agence nationale de sécurité du médicament et des produits de santé de tout risque de rupture de stock ou de toute rupture sur un médicament ou produit sans alternative thérapeutique disponible, dont elle assure l’exploitation, ainsi que de tout risque de rupture de stock ou de toute rupture sur un médicament ou produit dont elle assure l’exploitation, lié à un accroissement brutal et inattendu de la demande. Lorsque le médicament est utilisé dans une ou des pathologies graves dans lesquelles elle ne disposerait pas d’alternatives dispo-nibles sur le marché français, l’entre-prise apporte à l’agence sa collaboration à la mise en place de solutions alternatives permettant de couvrir ce besoin et des mesures d’accom-pagnement nécessaires. |
1° À la première phrase, après le mot : « informe », sont insérés les mots : « de manière motivée » ; 2° À la troisième phrase, les mots : « la notification doit avoir lieu » sont remplacés par les mots : « l’information de l’agence nationale de sécurité des médicaments et des produits de santé se fait, de manière motivée, » ; 3° Après la troisième phrase, il est inséré une phrase ainsi rédigée : « Dans tous les cas, l’entreprise pharmaceutique précise si la suspension ou l’arrêt de commercialisation du médicament est fondé sur l’un des motifs mentionnés aux 1° à 5° de l’article L. 5121-9. » |
|
Article 7 |
Article 7 | |
Art. L. 5121-1-2. – La pres-cription d’une spécialité pharmaceutique mentionne ses principes actifs, désignés par leur dénomination commune internationale recommandée par l’Organisation mondiale de la santé ou, à défaut, leur dénomination dans la pharmacopée européenne ou française. En l’absence de telles dénominations, elle mentionne leur dénomination commune usuelle. Elle peut également mentionner la dénomination de fantaisie de la spécialité. |
I. – À l’article L. 5121-1-2, les mots : « européenne ou française » sont supprimés et les mots : « la dénomination de fantaisie » sont remplacés par les mots : « le nom de fantaisie ». |
I. – À l’article L. 5121-1-2 du code de la santé publique dans sa rédaction résultant de l’article 19 de la loi n° 2011-2012 du 29 décembre 2011, relative au renforcement de la sécurité sanitaire du médicament et des produits de santé , les mots : … … fantaisie ». Amendement AS29 |
II. – Il est créé un article L. 5121-1-4 ainsi rédigé : |
||
« Art. L. 5121-1-4. – Lorsqu’elle est établie à la demande d’un patient en vue de l’utiliser dans un autre État membre de l’Union européenne, la prescription de l’un des médicaments mentionnés aux 6°, 14° et 15° de l’article L. 5121-1, à l’article L. 5121-3, ainsi qu’aux points a et d du 1 de l’article 2 du règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 concernant les médicaments de thérapie innovante, mentionne les principes actifs du médicament, désignés par leur dénomination commune internationale recommandée par l’Organisation mondiale de la santé ou, à défaut, par leur dénomination dans la pharmacopée, ainsi que le nom de marque et, le cas échéant, le nom de fantaisie du médicament prescrit. » |
||
III. – Après le premier alinéa de l’article L. 5121-11du même code, il est inséré un alinéa ainsi rédigé : | ||
Art. L. 5121-11. – L’autorisation de mise sur le marché prévue par l’article L. 5121-8 ne peut être attribuée pour un médicament dérivé du sang que lorsqu’il est préparé à partir de sang ou de composants du sang prélevés dans les conditions définies aux articles L. 1221-3 à L. 1221-7. |
||
« Dans des conditions déterminées par voie réglementaire, un médicament mentionné au premeir alinéa peut être marqué d’un pictogramme « Label éthique » indiquant qu’il est issu de sang ou de composants du sang prélevés dans les conditions définies aux articles L. 1221-3 à L. 1221-7. » Amendement AS10 | ||
Toutefois, à titre exceptionnel, une autorisation de mise sur le marché peut, par dérogation, être délivrée à un médicament préparé à partir de sang ou de composants de sang prélevés dans des conditions non conformes au second alinéa de l’article L. 1221-3 ou aux articles L. 1221-6 et L. 1221-7 si ce médicament apporte une amélioration en termes d’efficacité ou de sécurité thérapeutiques ou si des médicaments équivalents ne sont pas disponibles en quantité suffisante pour satisfaire les besoins sanitaires. Dans ce cas, l’autorisation de mise sur le marché est délivrée pour une durée de deux ans qui ne peut être renouvelée qu’en cas de persistance des conditions susnommées. |
||
IV. – L’article L. 5211-6 du même code est complété par un 7° ainsi rédigé : | ||
Art. L. 5211-6. – Sont détermi-nées, en tant que de besoin, par décret en Conseil d’État, les modalités d’application du présent titre, et notamment : 1° Les conditions auxquelles doivent satisfaire les organismes mentionnés au deuxième alinéa de l’article L. 5211-3 ; 2° Les modalités de la déclaration prévue à l’article L. 5211-3-1 ; 3° Les conditions dans lesquelles les dispositifs sur mesure peuvent être dispensés de la certification de conformité prévue à l’article L. 5211-3 ; 4° Les catégories de dispositifs et les procédures de certification qui leur sont applicables, ainsi que, le cas échéant, la durée pendant laquelle la certification est valable ;
5° Les catégories de dispositifs médicaux et les modalités de la communication prévues à l’article L. 5211-4, ainsi que les données devant être transmises à l’Agence nationale de sécurité du médicament et des produits de santé en application de cet article ; 6° Les modalités de l’évaluation des données cliniques des dispositifs médicaux mentionnées à l’article L. 5211-3-2. |
||
« 7° Les modalités de délivrance des dispositifs médicaux prescrits dans un autre État membre de l’Union européenne ainsi que les modalités de prescription des dispositifs médicaux en vue de leur délivrance dans un autre État membre. » Amendement AS2 |
Ordonnance 2012-1427 du 19 décembre 2012
Art. 1. – Après l'article L. 4211-1 du code de la santé publique, il est inséré un article L. 4211-1-1 ainsi rédigé :
« Art. L. 4211-1-1. – Les personnes se livrant à une activité de courtage de médicaments mentionnée à l'article L. 5124-19 ne sont pas soumises aux dispositions du présent titre. »
Art. 2. – Après l'article L. 5111-2 du code de la santé publique, il est ajouté un article L. 5111-3 ainsi rédigé :
« Art. L. 5111-3. – On entend par médicament falsifié tout médicament, tel que défini à l'article L. 5111-1, comportant une fausse présentation :
« 1° De son identité, y compris de son emballage et de son étiquetage, de son nom ou de sa composition s'agissant de n'importe lequel de ses composants, y compris les excipients, et du dosage de ces composants ;
« 2° De sa source, y compris de son fabricant, de son pays de fabrication, de son pays d'origine ou du titulaire de son autorisation de mise sur le marché ;
« 3° Ou de son historique, y compris des autorisations, des enregistrements et des documents relatifs aux circuits de distribution utilisés.
« La présente définition n'inclut pas les défauts de qualité non intentionnels. »
Art. 3. – L'article L. 5121-5 du code de la santé publique est ainsi modifié :
1° Au premier alinéa, les mots : « et la distribution en gros des médicaments » sont remplacés par les mots : « , la distribution en gros et l'activité de courtage de médicaments » ;
2° Au troisième alinéa, après le mot : « dispensation », sont insérés les mots : « , y compris par voie électronique, ».
Art. 4. – Au 3° de l'article L. 5121-20 du code de la santé publique, après le mot : « l'étiquetage, », est inséré le mot : « le conditionnement, ».
Art. 5. – À l'article L. 5124-1 du code de la santé publique, il est ajouté un dernier alinéa ainsi rédigé :
« Les personnes se livrant à une activité de courtage de médicaments mentionnée à l'article L. 5121-19 ne sont pas soumises aux dispositions du présent chapitre. »
Art. 6. – Après le chapitre IV du titre II du livre Ier de la cinquième partie du code de la santé publique, il est inséré un chapitre IV bis ainsi rédigé :
« Chapitre IV bis
« Courtage de médicaments
« Art. L. 5124-19. – On entend par activité de courtage de médicaments toute activité liée à la vente ou à l'achat de médicaments qui ne comprend pas de manipulation physique et qui consiste à négocier, indépendamment ou au nom d'une personne physique ou morale.
« Art. L. 5124-20. – Toute activité de courtage de médicaments effectuée par une personne située en France doit être déclarée auprès de l'Agence nationale de sécurité du médicament et des produits de santé.
« Les personnes exerçant des activités de courtage de médicaments veillent à ce que les médicaments faisant l'objet du courtage bénéficient d'une autorisation de mise sur le marché ou d'un enregistrement au titre des articles L. 5121-13 ou L. 5121-14-1.
« Les modalités de déclaration et d'exercice des personnes se livrant à l'activité de courtage sont prévues par décret en Conseil d'État. »
Art. 7. – I. – Après l'article L. 5122-6, il est inséré un article L. 5122-6-1 ainsi rédigé :
« Art. L. 5122-6-1. – Le commerce électronique de médicaments mentionné à l'article L. 5125-33 est soumis aux dispositions du présent chapitre. »
II. – Après le chapitre V du titre II du livre Ier de la cinquième partie du code de la santé publique, il est inséré un chapitre V bis ainsi rédigé :
« Chapitre V bis
« Commerce électronique de médicaments par une pharmacie d'officine
« Art. L. 5125-33. – On entend par commerce électronique de médicaments l'activité économique par laquelle le pharmacien propose ou assure à distance et par voie électronique la vente au détail et la dispensation au public des médicaments à usage humain et, à cet effet, fournit des informations de santé en ligne.
« L'activité de commerce électronique est réalisée à partir du site internet d'une officine de pharmacie.
« La création et l'exploitation d'un tel site sont exclusivement réservées aux pharmaciens suivants :
« 1° Pharmacien titulaire d'une officine ;
« 2° Pharmacien gérant d'une pharmacie mutualiste ou de secours minière, exclusivement pour leurs membres.
« Le pharmacien titulaire de l'officine ou gérant d'une pharmacie mutualiste ou de secours minière est responsable du contenu du site internet qu'il édite et des conditions dans lesquelles l'activité de commerce électronique de médicaments s'exerce.
« Les pharmaciens adjoints ayant reçu délégation du pharmacien d'officine peuvent participer à l'exploitation du site internet de l'officine de pharmacie.
« Les pharmaciens remplaçant de titulaires d'officine ou gérants d'officine après décès du titulaire peuvent exploiter le site internet de l'officine créé antérieurement par le titulaire de l'officine.
« Art. L. 5125-34. – Seuls peuvent faire l'objet de l'activité de commerce électronique les médicaments de médication officinale qui peuvent être présentés en accès direct au public en officine, ayant obtenu l'autorisation de mise sur le marché mentionnée à l'article L. 5121-8 ou un des enregistrements mentionnés aux articles L. 5121-13 et L. 5121-14-1.
« Art. L. 5125-35. – La création du site internet de commerce électronique de médicaments de l'officine de pharmacie prévu au troisième alinéa de l'article L. 5125-33 est subordonnée à l'existence de la licence mentionnée à l'article L. 5125-4 ou de la décision du ministre chargé de la santé mentionnée à l'article L. 5125-19 et à l'ouverture effective de la pharmacie.
« Art. L. 5125-36. – La création du site internet de commerce électronique de médicaments de l'officine de pharmacie est soumise à autorisation du directeur général de l'agence régionale de santé territorialement compétente. Le pharmacien informe de la création du site le conseil compétent de l'ordre des pharmaciens dont il relève.
« Art. L. 5125-37. – Dans le cadre d'un regroupement de plusieurs officines de pharmacie mentionné à l'article L. 5125-15, il ne peut être créé et exploité qu'un seul site internet rattaché à la licence issue du regroupement.
« La création du site internet issu du regroupement est soumise aux dispositions de l'article L. 5125-36.
« Ce site internet ne pourra être exploité que lorsque, le cas échéant, les sites internet de chacune des officines auront été fermés.
« Art. L. 5125-38. – La cessation d'activité de l'officine de pharmacie mentionnée à l'article L. 5125-7 entraîne la fermeture de son site internet.
« Art. L. 5125-39. – En cas de manquement aux règles applicables au commerce électronique prévues par les dispositions du présent chapitre et aux bonnes pratiques de dispensation mentionnées à l'article L. 5121-5 par l'un des pharmaciens mentionnés à l'article L. 5125-33, le directeur général de l'agence régionale de santé territorialement compétente peut, sauf en cas d'urgence, avoir mis en demeure, dans un délai qu'il fixe et qui ne peut être inférieur à huit jours, l'auteur du manquement de se conformer à ses prescriptions et de présenter ses observations :
« 1° Prononcer la fermeture temporaire du site internet de commerce électronique de médicaments pour une durée maximale de cinq mois ;
« 2° Prononcer une amende administrative à l'encontre de l'auteur du manquement et, le cas échéant, assortir cette amende d'une astreinte journalière qui ne peut être supérieure à 1 000 € par jour lorsque l'auteur de l'infraction ne s'est pas conformé à ses prescriptions à l'issue d'un délai fixé par une mise en demeure. Le montant de l'amende administrative ne peut être supérieur à 30 % du chiffre d'affaires réalisé par la pharmacie dans le cadre de l'activité de commerce électronique, dans la limite d'un million d'euros.
« Lorsqu'au terme de la durée de fermeture du site internet le pharmacien ne s'est pas mis en conformité avec les règles applicables, le directeur général de l'agence régionale de santé peut prononcer dans les mêmes conditions une nouvelle fermeture.
« L'agence régionale de santé informe le conseil de l'ordre compétent de la mise en œuvre de la procédure prévue au présent article.
« Art. L. 5125-40. – Une personne physique ou morale légalement habilitée à vendre des médicaments au public dans l'État membre de l'Union européenne dans laquelle elle est installée doit, dans le cadre d'une activité de commerce électronique de médicaments à destination d'une personne établie en France, respecter les dispositions de l'article L. 5125-34 ainsi que la législation applicable aux médicaments commercialisés en France.
« Art. L. 5125-41. – Les modalités d'application du présent chapitre, notamment les informations minimales que doivent contenir les sites internet de commerce électronique, sont déterminées par décret en Conseil d'État. »
Art. 8. – L'article L. 5138-1 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 5138-1. – Les activités de fabrication, d'importation et de distribution de substances actives, y compris en vue de l'exportation, ne peuvent être exercées que dans des établissements autorisés par l'Agence nationale de sécurité du médicament et des produits de santé.
« Toute activité de fabrication, d'importation ou de distribution d'excipients, y compris en vue de l'exportation, doit être déclarée auprès de l'agence. Toute modification des éléments constitutifs de la déclaration lui est immédiatement communiquée.
« Les conditions d'application du présent article sont déterminées par décret en Conseil d' État. »
Art. 9. – Le I de l'article L. 5138-2 du code de la santé publique est remplacé par les dispositions suivantes :
« I. – On entend par matières premières à usage pharmaceutique tous les composants des médicaments au sens de l'article L. 5111-1, c'est-à-dire :
« 1° La ou les substances actives. Est une substance active toute substance ou tout mélange de substances destiné à être utilisé pour la fabrication d'un médicament et qui, lorsqu'utilisé pour sa production, devient un composant actif de ce médicament exerçant une action pharmacologique, immunologique ou métabolique en vue de restaurer, corriger ou modifier des fonctions physiologiques, ou d'établir un diagnostic médical ;
« 2° Le ou les excipients. Est un excipient tout composant d'un médicament autre qu'une substance active et que les matériaux d'emballage. »
Art. 10. – I. – L'article L. 5138-3 du code de la santé publique est ainsi modifié :
1° Le premier alinéa est complété par une phrase ainsi rédigée :
« Les substances actives sont fabriquées et distribuées conformément à des bonnes pratiques dont les principes sont définis par le directeur général de l'Agence nationale de sécurité du médicament et des produits de santé, après avis de l'Agence nationale chargée de la sécurité sanitaire de l'alimentation, de l'environnement et du travail. » ;
2° Le deuxième alinéa est remplacé par cinq alinéas ainsi rédigés :
« Pour la fabrication de médicaments à usage humain, les établissements pharmaceutiques mentionnés à l'article L. 5124-1, les pharmacies à usage intérieur, les pharmacies d'officine ainsi que les médecins :
« 1° Vérifient la qualité et l'authenticité des matières premières qu'ils utilisent ;
« 2° Veillent à n'utiliser que des substances actives fabriquées et distribuées, y compris lorsqu'elles sont importées, conformément aux bonnes pratiques de fabrication et de distribution mentionnées au premier alinéa.
« Les établissements pharmaceutiques de fabrication de médicaments à usage humain mentionnés à l'article L. 5124-1 se conforment à l'obligation résultant du 2° notamment en réalisant, par eux-mêmes ou par l'intermédiaire d'un organisme tiers avec lequel ils concluent un contrat écrit, des audits sur les sites de fabrication et de distribution des substances actives.
« Ces mêmes établissements utilisent des excipients appropriés pour lesquels ils déterminent, sur la base d'une évaluation formalisée du risque conforme aux lignes directrices de la Commission européenne, les bonnes pratiques de fabrication adéquates. Cette évaluation du risque tient compte des exigences imposées par d'autres systèmes de qualité pertinents, de la source et de l'utilisation prévue de ces excipients, ainsi que de précédents cas de défaut de qualité. »
II. – Après l'article L. 5138-3, il est inséré un article L. 5138-3-1 ainsi rédigé :
« Art. L. 5138-3-1. – Pour la fabrication de médicaments à usage vétérinaire, les établissements pharmaceutiques mentionnés à l'article L. 5142-1, les vétérinaires, les pharmacies d'officine et les personnes autorisées à préparer des autovaccins à usage vétérinaire utilisent, en tant que matières premières à usage pharmaceutique, des substances actives répondant aux exigences du premier alinéa de l'article L. 5138-3. »
Art. 11. – L'article L. 5138-4 du code de la santé publique est ainsi modifié :
1° Au premier alinéa, après le mot : « conformité », sont insérés les mots : « , sauf pour les activités de distribution d'excipients » ;
2° Au dernier alinéa, après le mot : « l'agence », sont ajoutés les mots : « en coopération avec l'Agence européenne des médicaments ».
Art. 12. – Il est rétabli, dans le code de la santé publique, un article L. 5138-5 ainsi rédigé :
« Art. L. 5138-5. – Des substances actives ne peuvent être importées de pays tiers qu'à la condition d'avoir été fabriquées conformément à des normes de bonnes pratiques au moins équivalentes à celles fixées par l'Union européenne, et d'être accompagnées de documents définis par voie réglementaire attestant notamment le respect de telles normes. »
Art. 13. – Après l'article L. 5138-5 du code de la santé publique, il est ajouté un article L. 5138-6 ainsi rédigé :
« Art. L. 5138-6. – On entend par matière première à usage pharmaceutique falsifiée toute substance active ou tout excipient, dont l'usage pharmaceutique est établi, et comportant une présentation mensongère de son identité, y compris de son emballage et de son étiquetage, de son nom ou de sa composition, de son origine, y compris de son fabricant, de son pays de fabrication, ou de son historique, y compris des autorisations, des déclarations et des documents relatifs aux circuits de distribution utilisés. »
Art. 14. – L'article L. 5311-1 du code de la santé publique est ainsi modifié :
1° Au II, après les mots : « à la distribution en gros, », sont ajoutés les mots : « au courtage, » ;
2° Au troisième alinéa du III, après les mots : « produits de santé », sont ajoutés les mots : « , notamment sur les actions entreprises dans le domaine de la prévention et de la répression de la falsification des médicaments ».
Art. 15. – L'article L. 5312-4 du code de la santé publique est ainsi modifié :
1° La deuxième phrase de l'alinéa unique est supprimée ;
2° Il est ajouté deux alinéas ainsi rédigés :
« L'Agence nationale de sécurité du médicament et des produits de santé met en place un dispositif spécifique de veille et d'alerte visant à éviter, par la mise en œuvre de mesures d'information appropriées, que des médicaments susceptibles de présenter un danger pour la santé, en particulier lorsqu'ils sont soupçonnés d'être falsifiés ou d'être affectés de défauts de qualité, ne soient mis à la disposition des patients.
« Les mesures prises au titre des deux alinéas précédents et leur coût sont, le cas échéant, à la charge de la personne physique ou morale responsable de la mise sur le marché, de la mise en service ou de l'utilisation du ou des produits concernés. »
Art. 16. – I. – Le deuxième alinéa de l'article L. 5313-1 du code de la santé publique est complété par une phrase ainsi rédigée :
« Les inspections sont réalisées conformément aux bonnes pratiques définies par le directeur général de l'agence. »
II. – Au deuxième alinéa de l'article L. 5313-3 du même code, après le mot : « applicables », sont ajoutés les mots : « ainsi qu'aux bonnes pratiques mentionnées à l'article L. 5313-1 ».
Art. 17. – I. – Le chapitre Ier du titre II du livre IV de la cinquième partie du code de la santé publique est ainsi modifié :
1° Aux articles L. 5421-2 et L. 5421-3, après le mot : « commercialiser », sont insérés les mots : « , de réaliser l'activité de courtage » ;
2° Au chapitre Ier susmentionné, il est ajouté un article L. 5421-12 ainsi rédigé :
« Art. L. 5421-12 – Le fait de réaliser l'activité de courtage de médicaments mentionnée à l'article L. 5124-19, sans s'être déclaré auprès de l'Agence nationale de sécurité du médicament et des produits de santé en application et dans les conditions fixées par l'article L. 5124-20, est puni de deux ans d'emprisonnement et de 30 000 € d'amende. » ;
3° Après le chapitre Ier susmentionné, il est inséré un nouveau chapitre Ierbis ainsi rédigé :
« Chapitre Ierbis
« Médicaments falsifiés
« Art. L. 5421-13. – La fabrication, le courtage, la distribution, la publicité, l'offre de vente, la vente, l'importation, l'exportation de médicaments falsifiés définis à l'article L. 5111–3 sont punis de cinq ans d'emprisonnement et de 375 000 € d'amende.
« Les précédentes peines sont portées à sept ans d'emprisonnement et 750 000 € d'amende lorsque :
« 1° Le médicament falsifié est dangereux pour la santé de l'homme ;
« 2° Les délits prévus au premier alinéa ont été commis par des établissements pharmaceutiques autorisés conformément à l'article L. 5124-3, les courtiers déclarés conformément à l'article L. 5124-20, les pharmacies d'officine titulaires de la licence mentionnées à l'article L. 5125-4 et les pharmaciens à usage intérieur mentionnés à l'article L. 5126-5 du même code ;
« 3° Ces mêmes délits ont été commis en bande organisée ;
« 4° Les délits de publicité, offre de vente ou vente de médicaments falsifiés ont été commis sur un réseau de télécommunication à destination d'un public non déterminé.
« Art. L. 5421-14. – Sont punis de trois ans d'emprisonnement et de 75 000 € d'amende ceux qui, sans motif légitime, sont trouvés détenteurs de médicaments falsifiés.
« Lorsque le médicament falsifié est dangereux pour la santé de l'homme, les peines sont portées à cinq ans d'emprisonnement et à 375 000 € d'amende.
« Art. L. 5421-15. – La tentative des délits prévus à l'article L. 5421-13 est punie des mêmes peines. »
II. – Après le chapitre VII du titre III du livre IV de la cinquième partie du code de la santé publique, il est inséré un chapitre VIII ainsi rédigé :
« Chapitre VIII
« Matières premières à usage pharmaceutique
« Art. L. 5438-1. – Constitue un manquement soumis à sanction financière :
« 1° Le fait pour les fabricants, importateurs, distributeurs de substances actives de ne pas se conformer aux bonnes pratiques de fabrication et de distribution mentionnées au premier alinéa de l'article L. 5138-3 ;
« 2° Le fait pour tout établissement pharmaceutique mentionné à l'article L. 5124-1, pour les pharmacies d'officine, les pharmacies à usage intérieur, les médecins de ne pas s'assurer de la conformité des substances actives qu'ils utilisent aux bonnes pratiques de fabrication et de distribution, et, pour les établissements pharmaceutiques, de ne pas réaliser ou faire réaliser des audits pour s'en assurer sur les sites de fabrication et de distribution des substances actives ;
« 3° Le fait pour tout établissement pharmaceutique mentionné à l'article L. 5124-1 de ne pas déterminer les bonnes pratiques applicables à la fabrication d'excipients en réalisant une évaluation formalisée du risque et de ne pas s'assurer de leur respect.
« Art. L. 5438-2. – L'Agence nationale de sécurité du médicament et des produits de santé peut prononcer une amende administrative à l'encontre de l'auteur d'un manquement mentionné à l'article L. 5438-1. Elle peut assortir cette amende d'une astreinte journalière qui ne peut être supérieure à 2 500 € par jour lorsque l'auteur du manquement ne s'est pas conformé à ses prescriptions à l'issue du délai fixé par une mise en demeure.
« Le montant de l'amende prononcée pour les manquements mentionnés à l'article L. 5438-1 ne peut être supérieur à 10 % du chiffre d'affaires réalisé, dans la limite d'un million d'euros.
« Art. L. 5438-3. – Le fait pour le fabricant, l'importateur ou le distributeur de substances actives telles que définies par l'article L. 5138-2 d'exercer son activité sans y avoir été autorisé par l'Agence nationale de sécurité du médicament et des produits de santé en application et dans les conditions fixées par l'article L. 5138-1 est puni de deux ans d'emprisonnement et de 30 000 € d'amende.
« Art. L. 5438-4. – La fabrication, le courtage, la distribution, la publicité, l'offre de vente, la vente, l'importation, l'exportation, l'achat de matières premières à usage pharmaceutique falsifiées définies à l'article L. 5138-6 sont punis de cinq ans d'emprisonnement et de 375 000 € d'amende.
« Ces peines sont portées à sept ans d'emprisonnement et 750 000 € d'amende lorsque :
« 1° Ces matières premières sont dangereuses pour la santé de l'homme ;
« 2° Les délits prévus au premier alinéa ont été commis par des fabricants, importateurs, distributeurs autorisés ou déclarés en application de l'article L. 5138-1 ou par les établissements pharmaceutiques autorisés conformément à l'article L. 5124-3 ;
« 3° Ces mêmes délits ont été commis en bande organisée ;
« 4° Les délits de publicité, d'offre de vente et de vente de matière première à usage pharmaceutique falsifiées ont été commis sur un réseau de télécommunication à destination d'un public non déterminé.
« Art. L. 5438-5. – Sont punis de trois ans d'emprisonnement et de 75 000 € d'amende ceux qui, sans motif légitime, sont trouvés détenteurs de matières premières à usage pharmaceutique falsifiées.
« Lorsque la matière première à usage pharmaceutique falsifiée est dangereuse pour la santé de l'homme, les peines sont portées à cinq ans d'emprisonnement et à 375 000 € d'amende.
« Art. L. 5438-6. – La tentative des délits prévus à l'article L. 5438-2 est punie des mêmes peines.
« Art. L. 5438-7. – Les personnes morales déclarées pénalement responsables, dans les conditions prévues à l'article 121-2 du code pénal, des infractions prévues au présent chapitre encourent, outre l'amende suivant les modalités prévues à l'article 131-38 du même code, les peines prévues aux 2° à 9° de l'article 131-39 de ce code. »
Art. 18. – Au 4° de l'article L. 4211-1 du code de la santé publique, après les mots : « la vente au détail », sont ajoutés les mots : « , y compris par internet, ».
Art 19. – L'article L. 213-3 du code de la consommation est ainsi modifié :
1° Au 1°, les mots : « des substances médicamenteuses, » sont supprimés ;
2° Le 3° est abrogé ;
3° Au sixième alinéa, les mots : « ou si la substance médicamenteuse falsifiée » sont supprimés.
Art. 20. – L'article L. 213-4 du code de la consommation est ainsi modifié :
1° Le 3° est abrogé ;
2° Au sixième alinéa, les mots : « ou si la substance médicamenteuse falsifiée » sont supprimés.
Art. 21. – Au troisième alinéa de l'article L. 213-5 du code de la consommation, après la référence : « L. 5421-6-1 », sont ajoutées les références : « L. 5421-13, L. 5421-14, L. 5421-15, » et, après la référence : « L. 5431-7 », sont ajoutées les références : « L. 5438-3, L. 5438-4, L. 5438-5, L. 5438-6 ».
Art. 22. – Les articles 2, 3, 8, 9, 10, 13, 15, 16, 17, à l'exception des 1° et 2°, et 18 de la présente ordonnance, sont applicables à Wallis-et-Futuna, sous réserve des adaptations suivantes :
1° L'article L. 5521-7 du code de la santé publique est complété par les alinéas suivants :
« 4° Pour son application à Wallis-et-Futuna, l'article L. 5138-3 est ainsi modifié :
« a) Au deuxième alinéa, les mots : « les établissements pharmaceutiques mentionnés à l'article L. 5124-1, les pharmacies à usage intérieur, les pharmacies d'officine » sont remplacés par les mots : « la pharmacie de l'agence de santé » ;
« b) Au sixième alinéa, les mots : « conforme aux lignes directrices de la Commission européenne » sont supprimés ;
« 5° Le deuxième alinéa de l'article L. 5138-4 n'est pas applicable à Wallis-et-Futuna. » ;
2° L'article L. 5524-1 du code de la santé publique est ainsi modifié :
a) Le 1° est complété par les références : « L. 5421-13, L. 5421-14 et L. 5421-15 » ;
b) Les 9°, 10° et 11° deviennent respectivement les 10°, 11° et 12°, et il est inséré un 9° ainsi rédigé :
« 9° Les articles L. 5438-1 à L. 5438-7 ; ».
Art. 23. – I. – Les dispositions de l'article L. 5138-1 relatives à l'autorisation des activités de fabrication, d'importation ou de distribution de substances actives entrent en vigueur le 1er avril 2013. Les personnes exerçant régulièrement ces activités à la date de la publication de la présente ordonnance peuvent les poursuivre jusqu'à l'intervention de la décision de l'Agence nationale de sécurité du médicament et des produits de santé sur leur demande d'autorisation.
II. – Les pharmaciens aux 1° et 2° de l'article L. 5125-33 ayant déjà créé, à la date de la publication de la présente ordonnance, un site internet proposant des médicaments à la vente doivent déposer au plus tard le 1er mars 2013 la demande d'autorisation mentionnée à l'article L. 5125-36 du code de la santé publique. À partir de cette date, ils se conforment aux dispositions du chapitre V bis du titre II du livre Ier de la cinquième partie du même code. Ils peuvent néanmoins poursuivre cette activité jusqu'à l'intervention de la décision du directeur général de l'agence régionale de santé sur leur demande d'autorisation.
Art. 24. – Le Premier ministre, la garde des sceaux, ministre de la justice, la ministre des affaires sociales et de la santé et le ministre des outre-mer sont responsables, chacun en ce qui le concerne, de l'application de la présente ordonnance, qui sera publiée au Journal officiel de la République française.
ANNEXE
LISTE DES PERSONNES AUDITIONNÉES
(par ordre chronologique)
Ø Union des opticiens – Mme Catherine de La Boulaye, présidente et M. Jean-François Marinacce, président délégué
Ø Fédération nationale opticiens de France (FNOF) – M. Alain Gerbel, président
Ø Syndicat des opticiens sous Enseigne (SYNOPE) – M. Christian Romeas, président et Mme Alexandra Duvauchelle, déléguée générale
Ø Syndicat National de l'Optique Mutualiste (SYNOM) – M. Kulmie Samantar délégué général
Ø Association française des opticiens par Internet (AFOI) – M. Marc Adamowicz
Ø Sensee – M. Alain Colin, directeur général et M. Aurélien Pozzana, conseil
Ø Académie française de l’ophtalmologie M. Jean-Bernard Rottier, représentant du syndicat national des ophtalmologistes de France (SNOF), membre du Conseil national de la Spécialité (CNS)/
Ø Secrétariat général des affaires européennes (SGAE) – Mmes Juliette Clavière, Véronique Fourquet, Catherine de Lombard et M. Cassandre Barret
Ø Ministère des affaires sociales et de la santé – Direction générale de l’offre de soins (DGOS) – M. Guy Boudet, Chef du bureau RH2 « exercice , déontologie et développement professionnel continu des professions de santé », Mme Carole Merle, adjointe au chef du bureau RH2 et Mme Marion Sauvage, chargée de mission pour les professions de la rééducation au bureau RH2
Ø Ministère des affaires sociales et de la santé – Direction générale de la santé – Mme Claire Zemp, juriste au service judirique, Mme Emmanuelle Barsky, adjointe au chef du bureau dispositifs médicaux et Mme Magalie Guegan, adjointe au chef du bureau du médicament
Ø Ministère des affaires sociales et de la santé – Direction des affaires juridiques – M. Baptiste Messmer, conseiller juridique
Ø Agence nationale de sécurité du médicament et des produits de santé (ANSM) – M. Dominique Maraninchi, directeur général, et Mme Carole Le Saulnier, directrice des affaires juridiques et réglementaires
Ø Fédération des entreprises de la beauté (FEBEA) – M. Alain Grangé-Cabane, président, et Mme Anne Dux, Directrice des Affaires Scientifiques et Réglementaires
Ø Ordre national des pharmaciens – M. Jean-Charles Rochard, secrétaire général
Ø Fédération des syndicats pharmaceutiques de France (FSPF) – M. Philippe Besset, vice-président, et M. Philippe Liebermann, chargé des Affaires européennes
Ø Union nationale des pharmacies de France (UNPF) – Mme Françoise Daligault, présidente et Mme Balbi, conseiller technique
Ø Union des syndicats de pharmaciens d’officine (USPO) – M. Gilles Bonnefond, président
Ø Les entreprises du médicament (LEEM) – Mme Blandine Fauran, directeur juridique
Ø Association française de l’industrie pharmaceutique pour une automédication responsable (AFIPA) – M. Pascal.Brossard, président et Mme Daphné Lecomte-Somaggio, déléguée générale
Ø Union fédérale des ostéopathes de France – M. Dominique Blanc, président et M. Armand Gersanois, vice-président
Ø Syndicat français des Ostéopathes – M. Philippe Sterlingot, président, M. Thibault Dubois, vice-président et M. Joël Moret-Bailly, professeur agrégé des universités, spécialiste en droit de la santé
Ø Association française de Chiropratique (AFC) – M. Philippe Fleuriau, président
Ø Syndicat national des artistes tatoueurs (SNAT) – M. Cyril Auville, président, Mme Karine Laroque, secrétaire, MM. Pascal Guignon, Vincent Daniel, fournisseurs de produits et matériel de tatouage, membres du SNAT, et Me Benjamin Mercier, avocat et membre du SNAT
1 Conseil d’État, 28 avril 2003, Fédération française des courtiers d’assurance et réassurance
2 Conseil d’État, 8 février 2007, Société ARCELOR Atlantique et Lorraine et autres
3 Décisions n°189,190 et 191 de la Commission administrative sur la sécurité sociale des travailleurs migrants du 18 juin 2003.
4 La gestion du recouvrement des créances européennes et internationales aujourd’hui confiée au CLEISS est transférée à la Caisse nationale d’assurance maladie des travailleurs salariés (CNAM-TS), à compter du 1er janvier 2015, par l’article 81 de la loi de financement de la sécurité sociale pour 2014.
5 Avis n° 13-A-24 du 19 décembre 2013 relatif au fonctionnement de la concurrence dans le secteur de la distribution du médicament à usage humain en ville
6 Les autres motifs sont la non-conformité de la composition aux substances autorisées par la législation européenne, la nocivité dans les conditions normales d’emploi, ou le défaut d’effet thérapeutique.
7 La date d’entrée en vigueur de l’article 16 paragraphe 3 alinéa 2 du règlement 1223/2009 a été fixée au 11 janvier 2013 afin de s’assurer que les produits cosmétiques contenant des nanomatériaux mis sur le marché à partir du 11 juillet 2013 respecteraient les exigences posées par le règlement, et notamment l’obligation de notification six mois avant la mise sur le marché.
8 () L’article 23 de l’ordonnance a fixé la période de transition pendant laquelle les pharmaciens ayant déjà créé un site internet ont pu se mettre en conformité avec les nouvelles dispositions, une demande d’autorisation ayant dû être déposée au plus tard le 1er mars 2013.
9 () Les médicaments de thérapie génique comportent un principe actif composé d’un acide nucléique recombinant administré en vue de réguler, de réparer, de remplacer, d’ajouter ou de supprimer une séquence génique.
10 () Les médicaments de thérapie cellulaire contiennent des cellules ayant fait l’objet d’une manipulation substantielle afin de modifier leurs caractéristiques, fonctions ou propriétés ou des cellules n’étant pas destinées à être utilisées pour la même fonction chez le donneur et le receveur.
11 () Les produits issus de l’ingénierie cellulaire contiennent des cellules ou tissus issus de l’ingénierie cellulaire ou tissulaire et qui possèdent des propriétés permettant de régénérer, réparer ou remplacer un tissu humain.
12 ( Ces médicaments combinés de thérapie innovante associent un ou plusieurs dispositifs médicaux à un médicament de thérapie cellulaire ou tissulaire.
13 () La pharmacopée est un référentiel réglementaire définissant les caractéristiques scientifiques des principes actifs.